Article Text

Statistics from Altmetric.com
Editor—Familial hypertrophic cardiomyopathy (FHC) is an autosomal dominant disease, which may afflict as many as 1 in 500 subjects.1 The disease is characterised by an unexplained local or general myocardial hypertrophy and by myocyte disarray.2 Molecular genetic studies have so far identified nine disease associated genes, all of which encode sarcomeric proteins. The two genes in which most mutations have been described are the β-myosin heavy chain (MYH7)3 and the myosin binding protein C (MYBPC3) genes,4 each of which may account for up to 30% of all familial cases. Mutations in α-tropomyosin (TPM1),5troponin T (TNNT2),5 6troponin I (TNNI3),6 cardiac α-actin (ACTC),7 titin (TTN),8 and the essential (MYL3) and the regulatory (MYL2) myosin light chain genes have also been associated with FHC.9 This pronounced genetic heterogeneity may be the principal cause of the phenotypic variability that is seen in FHC. Thus, mutations inTNNT2 seem to be associated with sudden death at a young age,10 11 whereas families with mutations in MYBPC3 are generally characterised by progressive hypertrophy and a late onset of clinical manifestation.12 13 Furthermore, it has been proposed that a certain rare form of hypertrophic cardiomyopathy (HCM), asymmetric septal hypertrophy predominantly confined to the midventricular region, known as the midventricular hypertrophy (MVH) phenotype, may be associated with mutations in the two myosin light chain genes.9 However, limited and contradictory clinical information is available on FHC caused by mutations in these genes.9 14
We have studied MYL2 andMYL3 in 68 consecutively collected FHC families from Denmark and in 130 probands from South Africa. We established the frequency of myosin light chain mutations and assessed whether mutations in these two genes do cause a distinct phenotype. Concurrently, we identified subjects with an MVH phenotype in the Danish families and investigated the segregation of this phenotype in their relatives. Subsequently, we screened the probands of these families, as well as those in which we identified mutations, irrespective of their clinical phenotype, for the presence of mutations in the coding regions ofMYH7,MYBPC3, TNNT2,TPM1, TNNI3, andACTC to assess the correlation between the MVH phenotype and the causal mutation.
Methods
DANISH PATIENTS
Sixty-eight families (total number of subjects 395) were consecutively enrolled in a study of FHC at the National University Hospital, Copenhagen. The probands all fulfilled conventional criteria for FHC, and all had a maximum left ventricular wall thickness of 15 mm.12 15 After informed consent was obtained (Local Science Ethics Committee, No KF V92213), patient and family histories were taken and physical examinations, 12 lead ECG, and two dimensional and M mode echocardiography were performed. The echocardiographic investigations included Doppler measurements of left ventricular outflow tract, as well as transmitral flow patterns and allowed identification of patients with the MVH phenotype, which was defined as asymmetrical septal hypertrophy predominantly confined to the midventricular region.9 16
The relatives of these probands, hereafter identified by the family code and pedigree number, were then also clinically investigated to establish segregation of the phenotype. The clinical diagnosis of FHC in relatives was based on (1) ECG criteria of pathological Q waves or signs of left ventricular hypertrophy17 18 and/or (2) echocardiography criteria of a maximum left ventricular wall thickness ⩾13 mm17 18 in the absence of evidence of other specific heart muscle disease.
SOUTH AFRICAN PATIENTS
One hundred and thirty consecutively collected probands of mixed or white ancestry were enrolled as part of an ongoing study at the University at Stellenbosch. Patients were clinically characterised as described elsewhere.19 20 Briefly, the echocardiographic diagnosis of HCM was made in the presence of a maximum ventricular wall thickness ⩾13 mm in the absence of evidence of other specific heart muscle disease.
Additionally, blood samples from a group of 150 persons born in Denmark were used as controls in the genetic analysis.
DNA ISOLATION
Genomic DNA was purified from blood samples obtained from all subjects using a QIA-amp DNA purification kit (Qiagen, Germany).
MUTATION DETECTION TECHNIQUES
Genomic DNA was PCR amplified using AmpliTaq polymerase (Applied Biosystems, Foster City, CA). All probands were screened for mutations in theMYL2 and MYL3genes. The primers used for analysis of theMYL2 and MYL3genes are listed in table 1.
Primers used for the amplification of MYL2 and MYL3 coding sequences
The three probands with mutations in MYL2(ZY, ZG, and T), as well as the two probands from families with a member with MVH (ZJ and XI), were screened for mutations in all coding exons from the FHC causing genes (MYH7, MYBPC3,TPM1, TNNI3,TNNT2, ACTC,MYL2, and MYL3), except in MYH7, where only exons 3-23 were screened. Amplification was performed using intronic primers. Primers and conditions for analysis of these eight FHC genes are essentially as previously described.3-7 9 21 Mutation screening was performed by single stranded conformation polymorphism/heteroduplex (SSCP/HD) analysis.21 22 Aberrant SSCP/HD patterns were further investigated by DNA sequencing using an ABI377 DNA sequencer (Applied Biosystems).
Results
DISEASE CAUSING MUTATIONS IN THE TWO POPULATIONS
Mutation screening of MYL2 andMYL3 showed three Danish probands (of families ZY, ZG, and T) with mutations inMYL2, two novel and one previously described,9 but none with mutations inMYL3, while no disease causing mutations were found in the South African probands. The clinical data of mutation carriers are given in table 2.
Clinical and echocardiographic data of mutation carriers in the three FHC families (ZY, ZG, and T) carrying MYL2 mutations
The MVH phenotype was present in three Danish families (ZG, ZJ, and XI), but this was not a constant finding in all affected members of these families. An MYBPC3 mutation and anMYH7 mutation were detected in the probands of families ZJ and XI, respectively.
POLYMORPHISMS IN THE TWO POPULATIONS
The Danish patients were all white, whereas the South African patients were of either white or of mixed ethnic descent. Mutation screening of the two populations showed unique, as well as common, polymorphisms. Among South African probands of both white and mixed ancestry, a unique MYL2 intron 1 (g2579C>T) polymorphism was detected, whereas MYL2polymorphisms identified in exon 3 (g6027T>C) and intron 4 (g8393G>A) were present in both the South African and the Danish probands as well as the Danish control group. The exonic polymorphism did not cause an amino acid change, while the intronic polymorphisms are not expected to affect splicing. In MYL3, three silent variants were identified: exon 1 (c69C>T), exon 3 (c261C>T), and exon 4 (c399C>T). The former was found in both population groups and the control group and the latter two were found only in South African probands of white or mixed ancestry. None of these variants gave rise to amino acid changes.
MYL2LYS103GLU AND IVS6-1 MUTATIONS IN FAMILY ZY
Mutation screening of MYL2 identified aberrant SSCP conformers in both exon 5 and exon 7 in the proband from family ZY (ZY:III.2) (fig 1). Subsequent sequencing showed an A>G transition at position g8486 in exon 5, which generated a glutamic acid substitution at the highly evolutionarily conserved lysine at codon 103 (Lys103Glu), while sequencing of exon 7 showed a G>C transversion in the acceptor splice site of intron 6, IVS6-1. Mutation screening of exons 5 and 7 in the rest of family ZY showed that the Lys103Glu substitution was maternally (ZY:II.2) and the IVS6-1 mutation paternally (ZY:II.1) derived. On the maternal side, one other family member carried the Lys103Glu mutation (ZY:II.4). Neither of these mutations was found in any of the other 197 probands or 150 controls. No mutations were detected when screening the other seven FHC genes in this family.
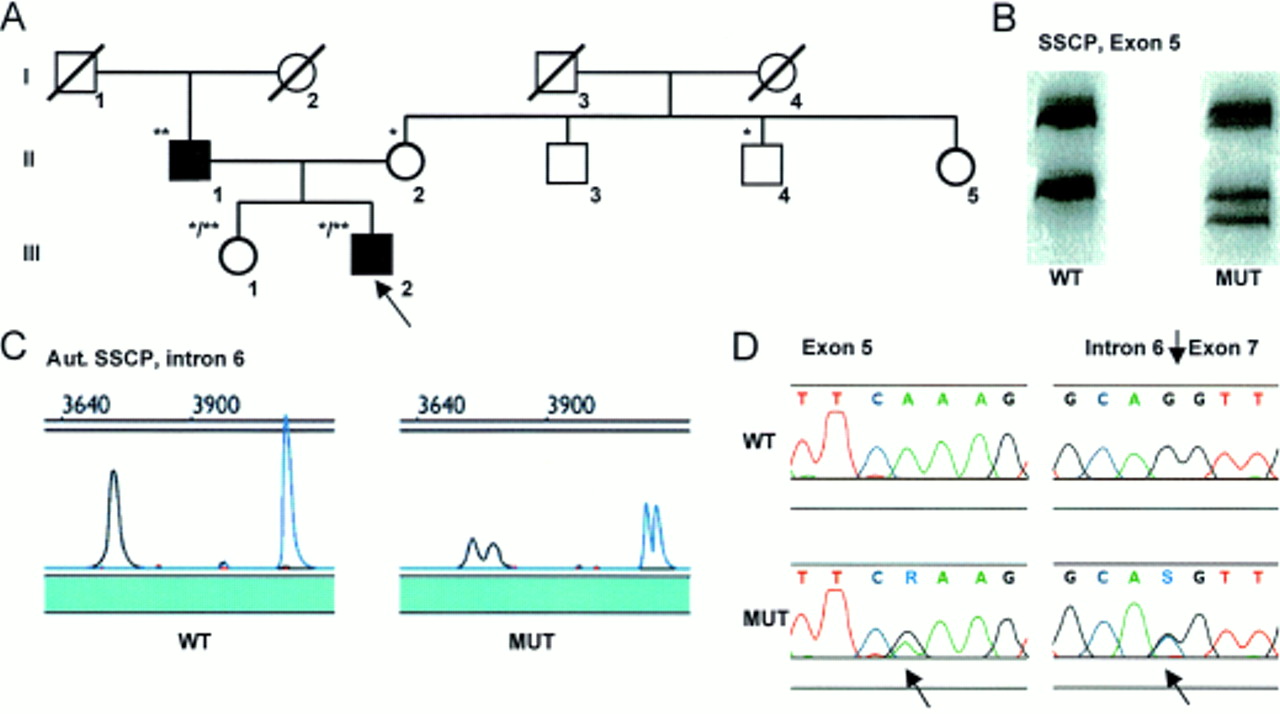
Pedigree of family ZY (A) with demonstration of the Lys103Glu MYL2 missense mutation by conventional SSCP/HD analysis (B) and of the IVS6-1 MYL2 splice site mutation by automated SSCP analysis (C), in both cases confirmed by DNA sequencing (D). The arrow denotes the proband. *Denotes heterozygosity for the Lys103Glu mutation, **denotes heterozygosity for the IVS6-1 mutation, and */** denotes compound heterozygosity. WT is the pattern seen in controls and MUT is the pattern seen in patients with a mutation.
The proband (ZY:III.2) was diagnosed with FHC at the age of 17 years. He had pronounced septal hypertrophy, predominantly confined to the proximal septum, and an inverted transmitral flow pattern indicating diastolic filling abnormalities (table 2). Despite the echocardiographic findings, he had only mild symptoms except for an episode of near syncope, angina, and severe dyspnoea during an attack of paroxysmal atrial fibrillation following intake of amphetamine-like drugs in order to lose weight. His sister (ZY:III.1), aged 42 years, also carrying both the missense and the splice site mutations, had no echocardiographic or ECG signs of hypertrophy, but experienced atypical chest discomfort. Subjects ZY:II.2 and ZY:II.4, carrying the Lys103Glu mutation, were asymptomatic and without ECG or echocardiographic signs of HCM (table 2). There was no family history of sudden death.
MYL2ALA13THR MUTATION IN FAMILY T
Mutation screening of exon 2 of theMYL2 gene in the proband (T:II.1) of family T (table 2) identified a G>A transition which causes the previously described FHC associated codon 13 Ala13Thr substitution.9Two other family members (T:II.2 and T:III.1) were heterozygous for this mutation, which was not present in either 150 controls or the other 197 probands. No other mutations were identified in this family in the additional seven FHC genes screened.
The proband (T:II.1), aged 42 years, suffered from exercise induced dyspnoea. He had pronounced septal hypertrophy, diastolic filling abnormalities, but no significantly increased left ventricular outflow tract (LVOT) gradient at rest or during Valsalva manoeuvre. However, during cycle ergonomic exercise there was a significant increase in the LVOT gradient, and he improved symptomatically following percutaneous transcoronary septal myocardial alcohol ablation. The mother (T:I.2) was diagnosed with FHC in later life and died at the age of 72 years. It was not possible to obtain tissue for genotyping. T:II.2 fulfilled the diagnostic criteria for FHC in her mid forties, whereas T:III.1, who was only 10 years old at the time of inclusion, did not. T:II.3, who was not a mutation carrier, had left ventricular hypertrophy, which seemed to relate to arterial hypertension and severe obesity. There was no family history of sudden death.
MYL2ASN47LYS MUTATION IN PROBAND ZG
A C>A transversion in exon 3 of MYL2, resulting in an Asn47Lys substitution, was identified in the proband (ZG:II.1) of family ZG (table 2). It was not possible to obtain blood samples from other family members. The mutation was not identified in the 150 healthy controls or in the other 197 probands. No other mutations were identified in this patient in the additional seven FHC genes screened.
Proband ZG was diagnosed with HCM at the age of 60 years, at which time (fig 2A, B), as well as during follow up (fig 2C, D), he had a high, relatively fixed midventricular flow gradient, as well as diastolic filling abnormalities. The midventricular flow gradient was caused not only by the pronounced septal hypertrophy, but also by a significant increase in the size of the papillary muscle apparatus (fig 2A, C). Interestingly, a marked progression in the septal hypertrophy, from 31 to 45 mm, was seen over two years from the age of 60 to 62 years (fig2). There was no family history of sudden death.

Echocardiographic data from patient ZG:II.1 with Asn47Lys MYL2 mutation showing the findings associated with the rare MVH phenotype. Panel A: parasternal long axis view in 1997 (60 years old) showing pronounced mid septal and papillary muscle apparatus hypertrophy. Panel B: apical two chamber view with left ventricular Doppler flow tracings showing the narrow mid ventricular flow (systole, with gradient) (yellow arrow). Panel C: parasternal long axis view in 1999 (62 years old) showing the increase of septal hypertrophy over two years. Panel D: apical four (two) chamber view showing diastolic in flow turbulence through the narrow mid ventricular region (yellow arrow). IVS, interventricular septum. PM, papillary muscle.
MUTATIONS IN TWO FAMILIES WITH MVH
In each of these two FHC families, the MVH phenotype was found in one member. In both families, other affected relatives displayed predominant hypertrophy of the proximal part of the septum. No myosin light chain mutations were found in either families. However, screening of the other six FHC causing genes identified a G>A transition at nucleotide g5166 of the MYBPC3 gene, resulting in an Asp228Asn substitution in proband ZJ, and a C>G transversion at g12739 of the MYH7 gene, causing an Asp778Glu substitution in proband XI, which was also present in his brother with the MVH phenotype.
Discussion
The observed frequency of MYL2mutations (4.4%) in this study of Danish patients is the second highest reported for this gene in any population to date9and justifies MYL2 mutation screening in the Danish HCM population. The frequency of MYL2mutations varies from 7.1% (3/42) in a French study9 to 0.8% (3/383) in an American study.9 In contrast, the absence of mutations in MYL2 in the South African population significantly reduces its precedence in mutation screening programmes in that population. The absence ofMYL3 mutations in both population groups studied confirms previous reports of the small contribution of this gene to the HCM disease load.9 While frequency studies may be influenced by differences in ethnic background, founder effects,19 referral bias, and differences in clinical inclusion criteria and mutation detection methods, the patients used in this study were included consecutively and screened in the same manner. However, the inclusion criteria for probands were less stringent in the South African population than in the Danish population (maximum left ventricular wall thickness of at least 13 mmv 15 mm), and this may partly explain why the MYL2 mutation frequency was lower in the former population.
-
Mutations in the genes, MYL2 and MYL3, coding for the sarcomeric myosin regulatory and essential light chains, respectively, cause familial hypertrophic cardiomyopathy (FHC).
-
We performed mutation screening of consecutively collected Danish (68) and South African (130) FHC probands to establish the frequency of MYL2 and MYL3 mutations. Subsequently, we undertook genotype-phenotype correlations to determine if these mutations are associated with the previously reported midventricular hypertrophy (MVH) phenotype. The coding regions of MYL2 and MYL3 were screened using single strand conformation polymorphism/heteroduplex analysis and mutations were identified by DNA sequencing.
-
In the Danish cohort, a novel splice site mutation (IVS6-1) and a novel missense mutation (N47K) were found in MYL2, in addition to one previously described mutation, but no MYL3 mutations were detected. No disease causing mutations were found in the South African cohort. Patients with MVH were identified in three Danish families, one carrying the MYL2 N47K mutation and the other two carrying mutations in the myosin light chain binding region of the β-myosin heavy chain gene or in the cardiac myosin binding protein C gene. Hence, MVH is neither unique to, nor invariably associated with, myosin light chain mutations.
-
The observed frequency of MYL2 FHC causing mutations (4.4%) in the Danish cohort warrants mutation screening of this gene in affected subjects in this population. The absence of light chain mutations in the South African probands corroborates reports of geographical variation in the frequency of FHC causing mutations. The absence of reported sudden death indicates that MYL2 mutations may be associated with a good prognosis.
Family ZY (fig 1) carries two MYL2mutations. Although the maternally derived Lys103Glu substitution occurs at a well conserved residue, codon 103 has not been included in the so far defined functional domains of the myosin regulatory light chain (RLC) (fig 3). In addition, as neither the mother (ZY:II.2) nor her sib (ZY:II.4), had a clinical phenotype compatible with FHC, it is probable that the Lys103Glu substitution that they carry is an extremely rare normal variant. Its absence in 150 controls supports this proposal; if, on the other hand, this is taken as evidence that Lys103Glu is, in fact, a disease causing mutation, its expression in family ZY would suggest it is associated with a low penetrance. In contrast, the paternally derived IVS6-1 splice site mutation, which is the first of this category described inMYL2, is likely to be the disease causing mutation, as clinical analysis of the proband's father (ZY:II.1) showed that he had pronounced proximal septal hypertrophy. Furthermore, the G>C tranversion affects the highly conserved AG sequence of the splice acceptor site.21 Neural network splice site prediction analysis predicts that this mutation results in a complete removal of the splice site, which may cause skipping of exon 6 and/or 7, or activation of a cryptic splice site 138 bp downstream from the substituted nucleotide. This would lead to truncation of the protein with loss of the functionally important MYH7binding site. Deletion of the carboxyl-terminus abolishes the phosphorylation mediated regulation of muscle contraction in smooth muscle.23 24 However, the mild phenotype of the proband's sister (ZY:III.1), who also has the splice site mutation, highlights the variable penetrance frequently associated with FHC causing mutations. The Ala13Thr MYL2mutation has previously been described as associated with the MVH phenotype of FHC,9 but this was not the case in family T. The novel Asn47Lys MYL2 mutation is found in the conserved Ca2+ binding domain of RLC, and we suggest that a changed affinity towards Ca2+ may be the pathophysiological mechanism of the mutation.

{kind=link}
{kind=link}
{kind=link}
Schematic drawing of the genomic structure of MYL2 marking the known FHC associated mutations and so far defined putative functional regions of the myosin regulatory light chain.
Phenotypically, most patients with HCM are of the classical type with predominantly proximal septal hypertrophy. Much more rare are the apical hypertrophy and the MVH phenotypes.16 The latter phenotype has in some cases been associated with mutations in the myosin light chains.9 However, others have also identified mutations in the light chains associated with the classical phenotype.14 We have found mutations inMYL2 associated with both classical (IVS6-1 and Ala13Thr) and the MVH form (Asn47Lys) (fig 2). The only common finding among the probands with MYL2mutations presented here are the family history of a good prognosis, that is, good survival (none of our families had members who had died unexpectedly), and late age of onset (children were free of symptoms of cardiac disease). However, it must be taken into account that the numbers of family members studied were small and more extensive investigations are necessary to confirm the good prognosis.
We also identified two additional families in which one affected family member had the MVH phenotype that was not caused by a mutation in either of the myosin light chains. One of these patients carried a mutation (Asp778Glu) in MYH7 in the region that has been characterised as the light chain binding region,9 while the other carried a mutation (Asp230Asn) inMYBPC3 in the amino (N)-proximal C1 domain. Interestingly, it has been speculated that the N-terminal region of myosin binding protein C may contribute to the regulation of muscle contractility, possibly in concert with the myosin regulatory light chain.25 In this region ofMYBPC3, one other missense mutation has previously been identified (Glu258Lys); however, this mutation has not been reported to be associated with the MVH phenotype. It is possible that mutations in this region may determine a hypercontractile state, which could induce hypertrophy directly.26
Acknowledgments
We gratefully acknowledge the technical assistance of Mads Dahm Johansen, Kirsten Lindboe, Jette Severinsen, and Jette Rasmussen. The work was supported by Director Emil C Hertz and wife Inger Hertz's foundation, the Danish Heart Foundation, the Danish Medical Research Council, Direktør Ib Henriksen's Foundation, Danish Medical Association Research Foundation, and Direktør Jacob Madsen and Hustru Olga Madsen's Foundation.