Article Text

Statistics from Altmetric.com
The autonomic nervous system has a craniosacral parasympathetic and a thoracolumbar sympathetic pathway (fig 1) and supplies every organ in the body. It influences localised organ function and also integrated processes that control vital functions such as arterial blood pressure and body temperature. There are specific neurotransmitters in each system that influence ganglionic and post-ganglionic function (fig 2).

The autonomic nervous system supply to various organs. Sweat glands (not included) are supplied by cholinergic fibres Reproduced from

The symptoms and signs of autonomic disease cover a wide spectrum (table 1) that vary depending upon the aetiology (tables 2 and 3). In some they are localised (table 4). Autonomic disease can result in underactivity or overactivity. Sympathetic adrenergic failure causes orthostatic (postural) hypotension and in the male ejaculatory failure, while sympathetic cholinergic failure results in anhidrosis; parasympathetic failure causes dilated pupils, a fixed heart rate, a sluggish urinary bladder, an atonic large bowel and, in the male, erectile failure. With autonomic hyperactivity, the reverse occurs. In some disorders, particularly in neurally mediated syncope, there may be a combination of effects, with bradycardia caused by parasympathetic activity and hypotension resulting from withdrawal of sympathetic activity. The history is of particular importance in the consideration and recognition of autonomic disease, and in separating dysfunction that may result from non-autonomic disorders.
Some clinical manifestations of autonomic dysfunction
Outline classification of autonomic diseases
Drugs, chemicals, poisons, and toxins causing autonomic dysfunction
Examples of localised autonomic disorders
CLINICAL FEATURES
General aspects
Autonomic disease may present at any age group; at birth in familial dysautonomia (Riley-Day syndrome), in teenage years in vasovagal syncope, and between the ages of 30–50 years in familial amyloid polyneuropathy (FAP). Neurodegenerative disorders affecting the autonomic nervous system often occur after the age of 50 years.
The majority of autonomic diseases are sporadic. Those genetically transmitted include the Riley-Day syndrome, an autosomal recessive disease where there is consanguinity in Ashkenazi Jews. There is an autosomal dominant trait in FAP. There often is a family history in vasovagal syncope, especially in those presenting below the age of 20 years. There may be a genetic predisposition in some disorders; drug induced autonomic disease may be caused by impaired metabolism or the production of toxic metabolites, as with perhexiline maleate neuropathy. A detailed history relating to drug usage, and chemical and toxin exposure (table 3) is always necessary. Drugs that usually have modest side effects may unmask autonomic deficits, such as hypotension caused by levodopa (L-dopa) in sympathetic failure. Autonomic disease may vary geographically; Chagas’ disease that affects millions is prevalent in South America where the blood sucking triatomine bugs transmit the causative organism, Trypanosoma cruzi.
Autonomic disease may affect only one organ or system (table 4) but may be an important feature of underlying disease. Thus, Horner’s syndrome, with mainly cosmetic effects, may be the harbinger of underlying non-autonomic disease (such as an apical lung neoplasm in Pancoast’s syndrome) or it may be an early sign of generalised autonomic failure. Gustatory sweating may follow surgery to the parotid gland (Frey’s syndrome) or be the result of diabetic autonomic neuropathy. In the generalised disorders as in multiple system atrophy (MSA), only a single system initially may be involved. Thus impotence in the male or urinary bladder dysfunction may pre-date other autonomic or neurological features (table 5). In parkinsonian patients, the early onset of autonomic dysfunction may lead to consideration of MSA. Alternatively, autonomic dysfunction in a longstanding parkinsonian patient may be the result of drugs. Autonomic neuropathy in diabetes is often associated with longstanding insulin dependence (type 1) and with a somatic neuropathy.
Some clinical manifestations in patients with primary autonomic failure: oropharyngeal dysphagia, urinary incontinence and respiratory features are less likely to occur in pure autonomic failure, and along with additional neurological deficits often are present in multiple system atrophy
The clinical features will now be considered under each major system.
Cardiovascular system
Orthostatic hypotension
The symptoms resulting from orthostatic (postural) hypotension often are the reason for requesting medical advice and may provide the initial clue to autonomic disease. Orthostatic hypotension is defined as a fall in blood pressure of 20 mm Hg systolic or 10 mm Hg diastolic on sitting, standing or during 60° head-up tilt (fig 3). This is expected to reduce the perfusion pressure of organs especially above heart level, such as the brain. Symptoms include dizziness, visual disturbances, and cognitive deficits (table 6) that may precede loss of consciousness.
Some of the symptoms resulting from orthostatic hypotension
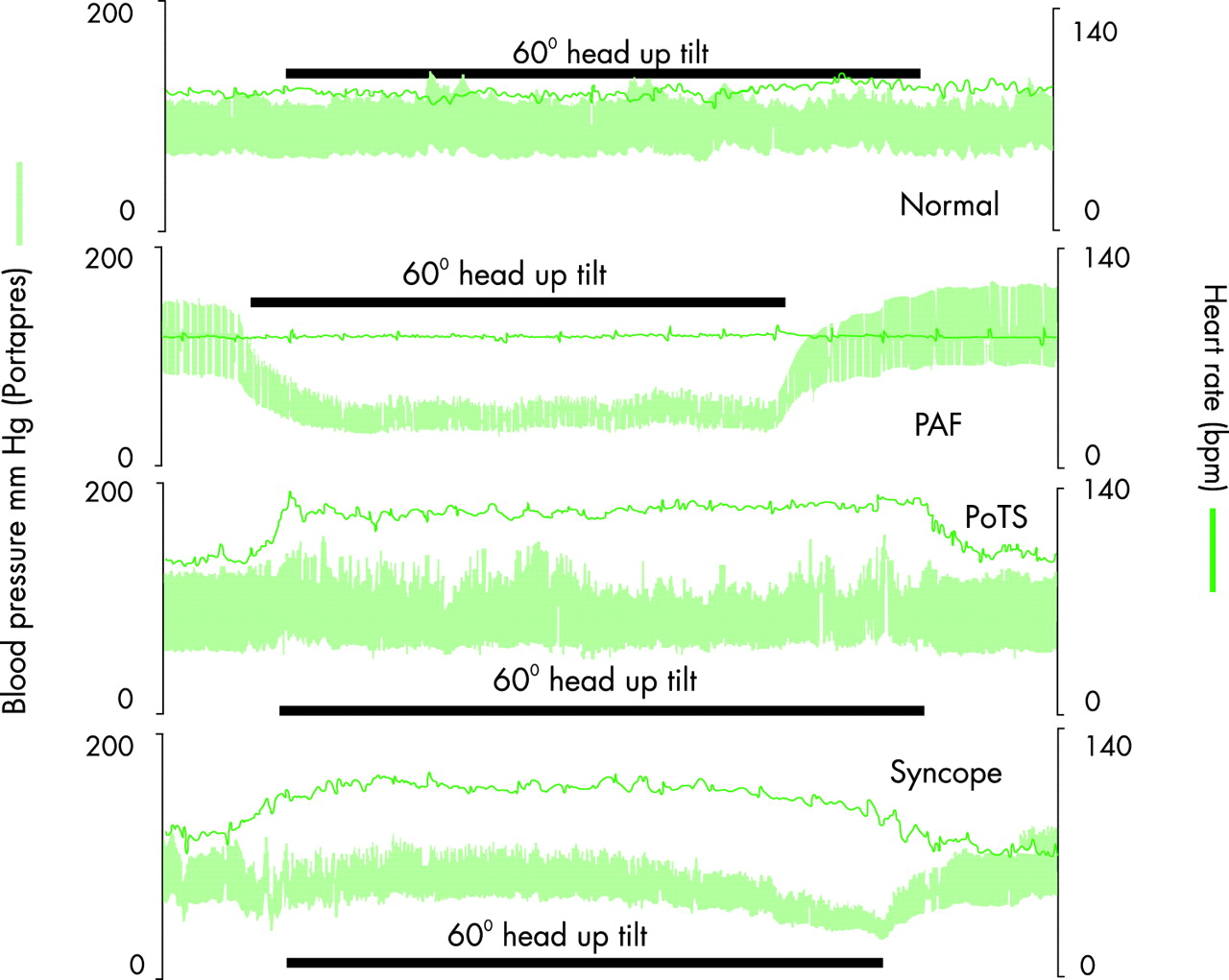
Blood pressure and heart rate measured non-invasively continuously before, during, and after 60° head-up tilt (by the Portapres II) in a normal subject and in subjects with three different autonomic disorders; with pure autonomic failure (PAF), postural tachycardia syndrome (PoTS), and vasovagal syncope. Reproduced from
The fall in blood pressure and associated symptoms during postural change may vary even within the same individual. If blood pressure falls precipitously, syncope may occur rapidly, as in a drop attack. This may result in injury. Occasionally, seizures may occur as a result of cerebral hypoxia. Many are at their worst in the early stages of their disorder. With time and frequent exposure to orthostatic hypotension, some tolerate a low cerebral perfusion pressure without symptoms, presumably because of improved cerebrovascular autoregulation. In some, a relatively small fall in blood pressure may induce cerebral hypoperfusion, especially in the presence of cerebrovascular disease.
A variety of symptoms result from hypoperfusion of other organs. Neck pain in a “coat hanger” distribution (affecting suboccipital and shoulder regions) differs from other types of neck pain by its presence when upright. It is relieved by sitting or lying flat, when the blood pressure recovers. It is probably caused by reduced perfusion of neck muscles that need to be tonically active to maintain the head upright. Activation of arm muscles, especially when upright (when reaching upwards, washing dishes, ironing clothes, and pushing a lawnmower), may increase cerebral symptoms of orthostatic hypotension by a subclavian steal-like syndrome by further reducing vertebral and thus brain stem blood flow. Central chest pain, suggestive of angina pectoris, may occur even in the young with normal coronary arteries; it may be caused by chest wall ischaemia. Lumbosacral and gluteal muscle discomfort, and in some calf claudication, also may occur. Some symptoms suggest spinal cord hypoperfusion.
Oliguria, especially during the day when upright, results from a reduction in renal perfusion pressure. This may be difficult to separate from retention of urine caused by urinary sphincter abnormalities, as in high spinal cord lesions. The reverse, nocturnal polyuria, occurs when supine, especially at night when blood pressure is restored or even elevated.
Non-specific symptoms include weakness, tiredness, and fatigue; in the elderly, falls may occur even without the other features of orthostatic hypotension.
A key component in the history is the relation between symptoms and head-up postural change. Symptoms may be more prominent with rapid head-up change, while getting out of bed in the morning, and on rising after eating a large meal. A variety of factors influence orthostatic hypotension and should be enquired about (table 7). Many recognise the association with head-up postural change and either sit down, lie flat, stoop or assume curious postures, such as squatting. These positions often prevent the fall in blood pressure or may even elevate blood pressure. Orthostatic hypotension can be considerably aggravated by drugs used to treat associated disease (L-dopa or insulin), to alleviate symptoms (nitrates) or to reverse organ failure (sildenafil). Drugs not usually associated with hypotension can lower blood pressure unduly in autonomic failure.
Factors influencing orthostatic hypotension
Syncope without orthostatic hypotension
Syncope has many causes (autonomic, cardiac, neurologic, and metabolic). Autonomic causes not resulting from orthostatic hypotension include neurally mediated syncope, an intermittent disorder with transient hypotension and bradycardia. Blood pressure falls because of sympathetic withdrawal, while heart rate falls as a result of increased vagal activity. This is more likely to occur when upright. Between attacks usually there are no autonomic abnormalities. The history of the syncopal attack and its recovery often separates these disorders from other neurological diseases, such as epilepsy. Recovery on lying flat usually is rapid, as this restores blood pressure and cerebral perfusion. Tongue biting does not occur. In some, convulsions may result from hypoxia, especially if the subject is not laid flat and blood pressure recovery is delayed. Urinary incontinence may occur occasionally.
In young and otherwise healthy individuals with preserved autonomic reflexes, a common cause is vasovagal syncope, also known as “common faints” or “emotional syncope”. Provoking factors include fear and pain. Common precipitants include venepuncture; the sight or even mention of a needle may induce an episode in needle phobia. This may represent abnormal conditioning of cerebral autonomic centres. Other factors include standing still, as occurs in children at school assembly or even fit young men on sentry duty, especially on a hot day. Nausea and other gastrointestinal upsets, probably through activation of visceral afferents, may be causative. Palpitations and sweating may occur in the pre-syncopal phase. In those with an adequate warning period, sitting or lying flat prevents syncopase. The reverse, standing or assumption of the upright position as on a tilt table, may provoke a response; the latter is the basis for the laboratory investigation of such disorders.
In the elderly, carotid sinus hypersensitivity may be a cause of falls. There may be a classical history of syncope induced while buttoning the collar, shaving or turning the head, when carotid afferents are stimulated. However, this may not be obtained, and falls and syncope of unknown aetiology should arouse suspicion of this disorder.
In situational syncope, various factors predispose the individual to syncope. These include induction of a Valsalva manouevre and hyperventilation as in weight lifters, trumpet blowers, and following paroxysms of coughing. In micturition syncope, hypotension results probably from the combination of vasodilatation caused by warmth or alcohol and straining during micturition (that raises intrathoracic pressure and induces a Valsalva manoeuvre), compounded by release of the pressor stimulus arising from a distended bladder while standing upright. Swallowing induced syncope may be associated with glossopharyngeal neuralgia.
Orthostatic intolerance with posturally induced tachycardia
Orthostatic intolerance may occur without orthostatic hypotension. In some there is a substantial rise in heart rate, of over 30 beats per minute, favouring the “postural tachycardia syndrome” (PoTS) or “neuropathic postural tachycardia syndrome” (NPTS). It predominantly affects women below the age of 50 years. Symptoms include pronounced dizziness on postural change or with modest exertion, usually without syncope. Their lives often are disrupted disproportionately. There usually are no features of generalised autonomic failure. Associated disorders include the hypermobility joint syndrome, chronic fatigue syndrome, mitral valve prolapse, and hyperventilation. Whether there is a relation to previously described psychosomatic disorders is unclear. A variant, more common in wartime, is DaCosta’s syndrome (soldier’s heart, or neurocirculatory asthenia), when dizziness and syncope on effort is accompanied by exhaustion, dyspnoea, headache, palpitations, and pain over the heart.
Prolonged bed rest and lack of exposure to gravitational forces, as in astronauts, also causes orthostatic intolerance.
Hypertension
Hypertension may cause few symptoms other than headache. Complications of severe hypertension include papilloedema, cerebral haemorrhage, aortic dissection, myocardial ischaemia and heart failure.
In high spinal cord lesions, severe paroxysmal hypertension may occur as part of autonomic dysreflexia, when an uninhibited increase in spinal sympathetic nervous activity is caused by contraction of the urinary bladder, irritation of the large bowel, noxious cutaneous stimulation, or skeletal muscle spasms. This may cause a throbbing or pounding headache, palpitations with bradycardia, sweating, and flushing over the face and neck; the limbs are cold because of peripheral vasoconstriction. In tetanus, hypertension may be precipitated by muscle spasms or tracheal suction in ventilated patients. Intermittent hypertension may occur in the Guillain-Barré syndrome, porphyria, posterior fossa tumours, and phaeochromocytoma, often without a clear precipitating cause. Sustained hypertension caused by increased sympathetic activity may occur in subarachnoid haemorrhage.
Hypertension in the supine position may complicate orthostatic hypotension in primary autonomic failure. The mechanisms include impaired baroreflex activity, adrenoceptor supersensitivity, an increase in central blood volume because of a fluid shift from the periphery, and the effects of drugs used to prevent orthostatic hypotension.
Heart rate disturbances
Bradycardia, along with hypertension, may occur in cerebral tumours and during autonomic dysreflexia in high spinal cord injuries. In the latter, the afferent and vagal efferent components of the baroreflex arc are intact, and the heart slows in an attempt to control the rise in blood pressure. In phaeochromocytoma, bradycardia with escape rhythms and atrioventricular dissociation may occur in response to a rapid rise in pressure.
Severe bradycardia can occur in artificially ventilated high cervical cord injuries with diaphragmatic paralysis. Their intact vagi are sensitive to hypoxia and stimuli such as tracheal suction induce bradycardia and cardiac arrest. The inability to increase sympathetic activity is likely to contribute. Similar responses may also occur in tetraplegics during general anaesthesia, especially when muscle paralysis followed by intubation is performed without atropine.
In neurally mediated syncope, severe bradycardia may occur in conjunction with hypotension. Syncope may occur despite preservation of heart rate by a cardiac demand pacemaker, because sympathetic withdrawal alone can cause substantial vasodilatation resulting in hypotension.
In diabetes mellitus, the presence of a cardiac vagal neuropathy may increase the likelihood of cardiorespiratory arrest during anaesthesia. Disorders of cardiac conduction are common in Chagas’ disease and occur in amyloidosis.
In PoTS, the tachycardia usually is associated with head-up postural change and exertion. Tachycardia caused by increased sympathetic discharge may occur along with hypertension in the Guillaine-Barré syndrome and in tetanus. In phaeochromocytoma, it results from autonomous catecholamine release and β adrenoceptor stimulation.
Facial and peripheral vascular changes
When blood pressure falls, in orthostatic hypotension or neurally mediated syncope, there usually is facial pallor, with an ashen appearance. There is prompt restoration of colour on assuming the supine position, when blood pressure rises. Facial pallor also may occur during an attack in phaeochromocytoma but usually is accompanied by sweating, headache, and hypertension. In chronic tetraplegia, hypertension during autonomic dysreflexia often is accompanied by flushing and sweating over the face and neck; the precise mechanisms are unknown. In Harlequin syndrome, there is vasodilatation and anhidrosis on one side of the face because of sympathetic impairment, with apparent sparing of the pupils. The lesion spares the first thoracic segment (from which oculomotor fibres often leave), but affects sympathetic fibres of the second and third thoracic roots. Raynaud’s phenomenon may occur in both PAF and MSA, for reasons not entirely clear. In the latter cold purplish blue hands and feet can be particularly troublesome. Livedo reticularis can accompany sympathetic overactivity, as in phaeochromocytoma. In erythromelagia there is limb discomfort with vascular changes. The precise reasons for the cutaneous, vascular, and sudomotor changes in reflex sympathetic dystrophy (chronic region pain disorder) remain debatable.
Sudomotor system
The eccrine glands are mainly concerned with temperature regulation. They are supplied by sympathetic cholinergic fibres, whereas the apocrine glands on the palms and soles are influenced by circulating substances, including catecholamines. Anhidrosis or hypohidrosis is common in primary autonomic failure, and differences in sweating may first be noticed during exposure to warm temperatures. Occasionally, hyperhidrosis in segmental areas may be the disconcerting presenting symptom, as a compensatory response to diminished sudomotor activity elsewhere. Anhidrosis may be congenital and occur without any other deficit. It may be an integral component of certain hereditary sensory and autonomic neuropathies, such as congenital insensitivity to pain with anhidrosis (type IV).
Localised or generalised anhidrosis, sometimes with compensatory hyperhidrosis, may be associated with the Holmes-Adie syndrome (Ross’ syndrome). In spinal cord injuries, there often is a band of hyperhidrosis above the lesion with anhidrosis below. During autonomic dysreflexia in high lesions sweating occurs mainly over the face and neck. Facial and trunkal hyperhidrosis may occur in Parkinson’s disease. Hyperhidrosis may occur intermittently in phaeochromocytoma and accompany hypertension in tetanus.
Localised hyperhidrosis over the face and neck caused by food (gustatory sweating) can be socially distressing. It occurs in diabetes mellitus or after parotid surgery, as a result of aberrant connections between nerve fibres supplying the salivary and sweat glands. Minimally invasive endoscopic techniques for sympathectomy often are successful in reducing axillary and palmar hyperhidrosis, but some develop troublesome compensatory hyperhidrosis over the trunk and lower limbs; the mechanisms are unclear.
Hypothermia may occur in hypothalamic disorders and in the elderly, in whom such lesions have been postulated. In high spinal injuries, especially in the early phases, the absence of “shivering thermogenesis” and an inability to vasoconstrict and thus prevent heat loss can readily result in hypothermia. Hypothermia may be missed if only oral temperature is recorded without a low reading thermometer; measurement of core (tympanic or rectal) temperature is essential.
Hyperpyrexia may be a problem in patients with anhidrosis exposed to a high ambient temperature. Heat also increases vasodilatation and can enhance orthostatic hypotension leading to collapse.
Alimentary system
Reduced salivation and a dry mouth (xerostomia) may occur in autonomic disease, especially in acute dysautonomias and in pure cholinergic dysautonomia. It may result in dysphagia when eating dry food. The lower two thirds of the oesophagus contains smooth muscle with an autonomic innervation, and autonomic diseases affecting these pathways may cause dysphagia. Oropharyngeal dysphagia is unusual in PAF, but often occurs in the later stages of MSA when it can result in aspiration. The oesophagus often is involved in Chagas’ disease, with achalasia and megaoesophagus causing vomiting. Gastroparesis in diabetes mellitus may cause abdominal distension and vomiting of undigested food.
Constipation is common in primary autonomic failure. Diarrhoea, which may be caused by overflow, may also occur. Diarrhoea, especially at night, can be a distressing problem in diabetes mellitus; the reasons include incomplete digestion, altered bowel flora, and abnormal motility.
Kidneys and urinary tract
Nocturnal polyuria is common in primary autonomic failure. The causes include restitution of blood pressure sometimes to raised levels while supine, with redistribution of blood from the peripheral into the central compartment and alteration in release of hormones (for example, renin, aldosterone, and atrial natriuretic peptide) that influence salt and water handling. In MSA where there is additional autonomic impairment of bladder and sphincter control, nocturia can be particularly troublesome. In the day, the low level of blood pressure when upright is likely to cause oliguria.
Autonomic disease may result in urinary frequency, urgency, incontinence, or retention. Loss of sacral parasympathetic function, as in the early phase of spinal cord injury, causes an atonic bladder with urinary retention, whereas recovery of isolated spinal cord function results in a neurogenic bladder. Dyssynergia, with detrusor contraction but without sphincter relaxation, causes autonomic dysreflexia. Urinary reflux predisposes to renal damage, especially in the presence of infection. In primary autonomic failure, urinary symptoms initially may be attributed in older men to prostatic hypertrophy and in women to pelvic muscle weakness, especially in those who are multiparous. In MSA, surgery in suspected prostate enlargement usually is of no benefit. The use of drugs with anticholinergic effects may unmask urinary bladder dysfunction in autonomic failure.
Infection is common when bladder dysfunction causes urine stasis. Some patients, such as with spinal injuries, are prone to urinary calculi, especially when immobility increases calcium excretion.
Sexual function
In the male, impotence may result from failure of erection, which is dependent on the parasympathetic system. Ejaculation is controlled by the sympathetic system. Retrograde ejaculation may occur, especially if there are urinary sphincter abnormalities. It may be difficult to dissociate the effects of increasing age, systemic illness, and depression from organic causes of impotence. The effect of drugs needs consideration. The 5 hydroxytryptamine (5-HT) uptake inhibitor, fluoxetine, prolongs ejaculation. Others normally not considered to have autonomic side effects, such as thiazides used in hypertension, may cause impotence.
Priapism caused by abnormal spinal reflexes may occur in patients with spinal cord lesions. In women, autonomic impairment does not appear directly to affect sexual function, although this has been inadequately studied.
Eye and lacrimal glands
The non-striated component of the levator palpebra superioris (Müller’s muscle) is innervated by sympathetic fibres, and mild ptosis is part of Horner’s syndrome. If the lesion is bilateral, as in high spinal cord transection, this is difficult to detect. A variety of pupillary abnormalities may occur in autonomic disease: miosis in Horner’s syndrome and dilated myotonic pupils in Holmes-Adie syndrome. Night vision may be impaired in sympathetically denervated pupils. There is reduced tolerance to sunlight when pupils are dilated because of parasympathetic failure. The ciliary muscle is innervated by parasympathetic nerves and blurred vision caused by cycloplegia may result with disease or anticholinergic drugs; the latter also may raise intraocular pressure and contribute to glaucoma.
Impaired lacrimal production may occur in primary autonomic failure, sometimes as part of an apparent sicca or Sjögren’s syndrome, along with diminished salivary secretion. Excessive and inappropriate lacrimation occurs in the crocodile tears syndrome (gusto-lacrimal reflex).
Respiratory system
Involuntary inspiratory sighs, stridor, and snoring of recent onset are more frequent in MSA than in Parkinson’s disease. Stridor results from weakness of the cricoarytenoid muscles, the main laryngeal abductors. Nocturnal apnoea, that occurs in the later stages of the disorder, is caused by involvement of brainstem respiratory centres.
Abnormal responses following activation of reflexes from the respiratory tract, such as during tracheal suction, may cause profound cardiovascular disturbances; in tetanus severe hypertension and tachycardia, while in high cervical cord transaction bradycardia and cardiac arrest, may occur.
Additional neurological involvement
In the parkinsonian forms of MSA, bradykinesia and rigidity with minimal tremor are more likely, in contrast to Parkinson’s disease. This causes difficulties in mobility, especially while turning in bed and changing direction. Facial expression is affected to a lesser degree than in Parkinson’s disease. There often is a response to antiparkinsonian agents in the early stages; side effects, such as orthostatic hypotension and motor refractoriness, are likely to occur as the disease progresses. In Parkinson’s disease with autonomic failure, extrapyramidal features often have been present for a long period and usually remain responsive to L-dopa treatment.
In the non-parkinsonian forms of MSA, cerebellar features predominate with an ataxic gait, intention tremor, scanning speech, and nystagmus. Ataxia may be difficult to separate from, or may be compounded by, unsteadiness caused by orthostatic hypotension. There may also be pyramidal involvement with increased tone, exaggerated tendon reflexes, and extensor plantar responses. A varying combination of extrapyramidal, cerebellar, and pyramidal features occurs in the mixed form of MSA. Sensory deficits are uncommon in MSA.
Patients with secondary autonomic failure have neurological features that are a part of, or a complication of, the primary disease. In diabetes mellitus, a somatic neuropathy often coexists with, or precedes, the autonomic neuropathy.
Psychological and psychiatric disturbances
Dementia is unusual in primary autonomic failure. In MSA, deficits in visuospatial organisation and visuomotor ability are similar to observations in Parkinson’s disease. The majority with MSA are not depressed, despite their disabilities and the probable deficit in central catecholamine concentrations; overall they have a normal affective state, especially when comparisons are made with Parkinson’s disease. In PAF there is no psychological disorder, but the absent autonomic responses may result in subtle deficits. They appear less emotional than normal subjects, and when compared to similarly disabled patients with Parkinson’s disease without autonomic failure, are less anxious. Cognitive function may transiently be affected when blood pressure falls below cerebral perfusion pressure limits; whether this affects certain tasks—for example, involving attention—is unclear.
Anxiety and tremulousness may occur in phaeochromocytoma. Psychological factors may contribute to vasovagal syncope (hence the term “emotional syncope”) and also in essential hyperhidrosis. Whether this is the cause, or result, of the autonomic condition can be difficult to dissect. Frank psychiatric disturbances may complicate conditions such as porphyria.
CLINICAL EXAMINATION
A detailed physical examination is essential and with the symptoms elicited may provide important clinical pointers towards autonomic disease. Features on general examination include dryness of skin, hyperhidrosis or cold hands in Raynaud’s. Measurement of blood pressure, both lying and sitting or standing is essential to determine if orthostatic hypotension is present, as is the pulse rate, especially in PoTS. A detailed neurological examination should include evaluation of pupillary function. The extent and distribution of the neurological abnormalities provide important clues to underlying central or peripheral autonomic disorders. Examination of other systems, as in hepatic disease or diabetes, is important for ascertaining accurately the underlying diagnosis and associated complications; and also for interpreting the results of autonomic tests, in the context of the associated disorder.
LABORATORY ASSESSMENT
The majority of investigations ideally are performed in autonomic laboratories. Such facilities should be available in major neuroscience centres. Screening tests predominantly are directed to evaluation of the cardiovascular autonomic nervous system (table 8). Additional tests may be needed. Laboratory investigation is for at least three purposes:
Outline of investigations in autonomic diseases
-
to determine if autonomic function is normal or abnormal
-
to evaluate, if an abnormality has been observed, the degree of autonomic dysfunction, with an emphasis on the site of lesion and the functional deficits
-
to ascertain if autonomic dysfunction is of the primary or secondary variety (table 2), as this determines the extent of further investigations, prognosis, and may modify management strategies.
In disorders such as neurally mediated syncope, testing may need to be designed around the individual patient and the circumstances associated with, or contributing to, the autonomic disorder. In generalised autonomic diseases, investigation of various systems may be required.
Cardiovascular system
A postural fall in blood pressure, if more than 20 mm Hg systolic, or less in the presence of symptoms, warrants further investigation. In the clinic this can be performed while lying down and then sitting or standing. In the laboratory head-up tilt to 60° often is used as the postural stimulus, especially when the neurological deficit or severe hypotension makes it difficult for the patient to stand upright. Blood pressure and heart rate can be accurately measured using non-invasive techniques, many of which are automated and provide a printout at preset intervals. In autonomic failure there may be considerable variability in the basal supine levels and also the postural fall in blood pressure; the greatest changes often occur in the morning, after a meal, and following physical exertion. There are many causes for orthostatic hypotension encountered in clinical practice, as outlined in patients with parkinsonian disorders (table 9). Non-neurogenic causes must be considered (table 10), especially as they worsen neurogenic orthostatic hypotension.
Orthostatic hypotension in parkinsonian disorders
Examples of non-neurogenic causes of orthostatic hypotension. In patients with autonomic failure these may enhance orthostatic hypotension considerably
Autonomic screening tests, in addition to head-up tilt testing, help determine the site and extent of the cardiovascular autonomic abnormality. The responses to Valsalva’s manoeuvre, during which intrathoracic pressure is raised to a maximum of 40 mm Hg, depend on the integrity of the entire baroreflex pathway. Changes in heart rate alone may provide a useful guide. Some patients, however, may raise mouth pressure without necessarily raising intrathoracic pressure, resulting in a falsely abnormal heart rate response. Stimuli that raise blood pressure, such as isometric exercise (by sustained hand grip for three minutes), the cold pressor test (immersing the hand in ice slush for 90 seconds), and mental arithmetic (using serial −7 or −17 subtraction), activate different afferent or central pathways, which then stimulate the sympathetic outflow. The heart rate responses to postural change, deep breathing (sinus arrhythmia), and hyperventilation assess the cardiac parasympathetic (vagus).
Additional investigations may be needed to determine factors causing or contributing to orthostatic hypotension and syncope. These include the responses to food ingestion, exercise, and carotid sinus massage. To assess postprandial hypotension, the cardiovascular responses to a balanced liquid meal containing carbohydrate, protein, and fat are measured while supine, with comparisons of the blood pressure response to head-up tilt before the meal and 45 minutes later. To evaluate exercise induced hypotension, responses are obtained during graded incremental supine exercise using a bicycle ergometer with measurement of postural responses before and after exercise. In suspected carotid sinus hypersensitivity, resuscitation facilities should be available because carotid massage may cause profound bradycardia or cardiac arrest (fig 4). Massage also should be performed during head-up tilt as hypotension may occur only in this position because of the greater dependence on sympathetic tone. Intermittent ambulatory blood pressure and heart rate recordings over a 24 hour period using small computerised lightweight devices are of particular value, especially at home, in determining the effects of various stimuli in daily life. However, it is essential in autonomic disease, in contrast to hypertensive patients, that appropriate protocols are followed and an accurate diary of events maintained to determine the effects of postural change, food, and exercise. The information is of value in determining the effects of treatment.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Continuous blood pressure and heart rate measured non-invasively (by Finapres) in a patient with falls of unknown aetiology. Left carotid sinus massage caused a fall in both heart rate and blood pressure. The findings indicate the mixed (cardio-inhibitory and vasodepressor) form of carotid sinus hypersensitivity. Reproduced from
Plasma catecholamine measurements are available in specialised laboratories and may be of value in certain disorders. Plasma noradrenaline (norepinephrine) provides a measure of sympathetic neural activity and plasma adrenaline (epinephrine) of adrenal medullary activity. In PAF, the supine basal concentrations of plasma noradrenaline are low (suggesting a distal lesion) compared with MSA, in whom supine values are often within the normal range. In both groups, there is an attenuation or lack of rise in plasma noradrenaline during head-up tilt, indicating impairment of sympathetic neural activity. In high spinal cord lesions, basal plasma noradrenaline and adrenaline concentrations are low and do not rise with postural change. There is, however, a rise (but only moderately above the basal values of normal subjects) during hypertension accompanying autonomic dysreflexia, which differs with paroxysmal hypertension caused by a phaeochromocytoma, when plasma noradrenaline or adrenaline concentrations usually are greatly elevated.
Extremely low or undetectable concentrations of plasma noradrenaline and adrenaline with raised plasma dopamine concentrations occur in sympathetic failure caused by deficiency of the enzyme dopamine beta-hydroxylase (DBH), which converts dopamine into noradrenaline.
In Addison’s disease with adrenocortical failure, a Synacthen test confirms the diagnosis; basal plasma renin concentrations are raised, whereas plasma aldosterone is low or absent. In diabetic autonomic neuropathy, there may be low concentrations of both plasma renin and aldosterone, which contribute to hyperkalaemia.
Muscle and skin sympathetic nervous activity can be recorded directly by percutaneous insertion of tungsten microelectrodes into the peroneal or median nerve. Muscle sympathetic activity is closely linked to the baroreceptor reflex, with a clear relationship to blood pressure. In high spinal cord lesions there is reduced baseline neural activity consistent with low basal plasma noradrenaline and blood pressure levels, because of the lack of transmission of tonic brainstem sympathetic activity. Increased nerve firing occurs in the Guillain-Barré syndrome, with hypertension and tachycardia. These microneurographic approaches have aided our understanding of the pathophysiological processes but are of limited clinical application, especially in the investigation of autonomic failure.
Pharmacological approaches determine the degree of sensitivity of different receptors and the functional integrity of sympathetic nerves and cardiac vagi. Some have value in the clinical situation. Repeat head-up tilt after stepwise intravenous atropine (to a maximum of 1800 μg), when the rate rises to 110 beats/min, helps determine the role of maintaining heart rate (such as by cardiac pacing), in the cardio-inhibitory forms of vasovagal syncope. A vasodepressor response without bradycardia post-atropine indicates that pacing is unlikely to be effective.
Certain pharmacological challenges, as with the α2 adrenoceptor agonist clonidine, provide information in different disorders. Basal plasma noradrenaline concentrations may be raised because of stress and other factors; in these situations, the central sympatholytic actions of clonidine suppress plasma noradrenaline values which does not occur with autonomous secretion in pheochromocytoma. Another central action of clonidine, through the hypothalamus and anterior pituitary, is stimulation of growth hormone release. Serum growth hormone concentrations rise in normal subjects and in pure autonomic failure (PAF) who have distal autonomic lesions; there is no response in MSA, in whom the lesions are central. The absent response in MSA is not caused by an inability to release growth hormone, as there is a growth hormone response to L-dopa. Thus, neuropharmacological challenge with clonidine separates the two disorders, MSA and PAF. Whether the clonidine growth hormone test will also aid distinction of parkinsonian forms of MSA from idiopathic Parkinson’s disease remains to be determined.
Advances in modern technology enable non-invasive measurement of cardiac function and blood flow in various regions. A variety of spectral analytic techniques assess cardiovascular function. Radionuclide 123-meta-iodo-benzylguanidine imaging assesses cardiac sympathetic innervation. Invasive techniques measure total body and regional noradrenaline spillover in the heart, splanchnic, and renal circulations and brain. These techniques have a role in the clinical research setting, and in due course some may be applied to the clinical investigation of cardiovascular autonomic function.
Sudomotor function
In the thermoregulatory sweat test, body temperature is raised by 1°C with a heat cradle or hot water bottles and a space blanket. This tests the integrity of central pathways, from the hypothalamus to the sweat glands. Sweating is assessed using powders, such as quinazarine or Ponceau red, which turn form a pale pink to a vivid purple red on exposure to moisture. In autonomic failure, the thermoregulatory sweating response is usually lost but this does not distinguish between central and peripheral lesions. In postganglionic lesions, the sudomotor and pilomotor response to intradermal acetylcholine also is lost. Methods to test this include the quantitative sudomotor axon reflex test. Most measure sweat production over a small area. Intradermal pilocarpine directly assesses the function of sweat glands. In DBH deficiency, sympathetic cholinergic function and sweating is preserved, indicating selective impairment of sympathetic noradrenergic function. In gustatory sweating, spicy foods, cheese, or substances containing tyramine are ingested to provoke sweating.
The sympathetic skin response (SSR) measures electrical potentials from electrodes on the foot and hand and indicate sympathetic cholinergic activity to sweat glands. The stimuli used are physiological (inspiratory gasps, loud noise, or touch) or electrical (median nerve stimulation). In peripheral autonomic diseases, such as PAF and pure cholinergic dysautonomia, the SSR is absent. In MSA with confirmed sympathetic adrenergic failure, a third have a recordable SSR. The SSR is absent below the level of lesion in complete spinal cord lesions.
Gastrointestinal tract
Video-cinefluoroscopy is of value in assessing swallowing and the presence of oropharyngeal dysphagia, especially in MSA patients who develop difficulties in deglutition, which enhance the tendency to aspiration pneumonia. A barium swallow, meal, and follow through are helpful in suspected upper gastrointestinal disorders; alternative investigation by endoscopy provides the opportunity for biopsy. Oesophageal manometry is of value in disorders of motility and oesophagogastric function. Several methods determine gastric motility non-invasively. When bacterial overgrowth is a suspected cause of diarrhoea, a therapeutic trial with broad spectrum antibiotics, such as neomycin or tetracycline, may be used along with investigations such as jejunal aspiration and the C14 glycocholate test. Helicobacter pylori infection is common in MSA and PAF and may contribute to gastric symptoms. Small bowel manometry and telemetric devices are of value in separating myopathic from neuropathic disorders of the gut.
Renal function and urinary tract
Nocturnal polyuria can be assessed by day and night urine volumes. Measurement of urine osmolarity and plasma sodium and potassium may be needed. When the urinary bladder is involved, an intravenous pyelogram and micturating cystometrogram may be needed. Urodynamic measurements define function of the bladder musculature and sphincter mechanisms. They may differentiate Parkinson’s disease from MSA; in the former, detrusor hyperreflexia may be present, whereas in MSA, there usually is a combination of both detrusor hyperreflexia and stress incontinence caused by a weak urethral sphincter. Measurement of postmicturition residual volume (for example, by ultrasound) is of importance. It may be high when the bladder is involved, as in MSA, and may result in urinary infection, which should be detected early and promptly treated.
Urethral sphincter electromyography provides an analysis of motor units affected when there is degeneration of Onuf’s nucleus in the sacral cord. This results in sphincter denervation with subsequent reinnervation. Electromyography indicates an increase in amplitude and duration of individual motor units, which often are polyphasic. This combination of denervation and reinnervation is present in the various forms of MSA, unlike Parkinson’s disease; similar changes occur in the anal sphincter.
Respiratory system
Sleep studies are needed when apnoeic episodes and stridor are present. Indirect and direct laryngoscopy detect laryngeal abductor paresis.
Eye and lacrimal glands
Various physiological and pharmacological tests help determine sympathetic or parasympathetic involvement of pupils. Dilute cholinomimetics assess pupillary sensitivity, that is enhanced with denervation in the Holmes-Adie pupil. Lacrimal secretion can be tested by Schirmer’s test, and damage from deficient secretion can be assessed using Rose Bengal instillation followed by slit lamp examination.
Miscellaneous
Additional investigations may be needed to determine the cause of autonomic disease, the underlying disorder or associated complications. Computed tomographic scans and magnetic resonance imaging of the brain help in assessing basal ganglia, cerebellar, and brainstem involvement in the primary autonomic failure syndromes, especially in MSA. In suspected peripheral nerve involvement, electrophysiological studies together with sural nerve biopsy are indicated. In amyloidosis, a rectal or renal biopsy may be diagnostic. In FAP, genetic studies confirm the diagnosis, the mutation, and also help determine which family members are at risk. To exclude adrenal insufficiency, a short or long Synacthen test should be performed.
In localised lesions, specific investigations to determine the cause may be warranted. In Horner’s syndrome this may include neuro-imaging to exclude a midbrain or medullary haemorrhage, radiography and bronchoscopy to exclude an apical bronchial neoplasm, and carotid artery angiography to assess lesions of the internal carotid artery.
REFERENCES AND FURTHER READING
Footnotes
-
↵* Recommended.