Article Text

Abstract
Transient neonatal diabetes (TND) is a rare but distinct type of diabetes. Classically, neonates present with growth retardation and diabetes in the first week of life. Apparent remission occurs by 3 months but there is a tendency for children to develop diabetes in later life. Evidence suggests it is the result of overexpression of an imprinted and paternally expressed gene/s within the TND critical region at 6q24. Two imprinted genes, ZAC (zinc finger protein associated with apoptosis and cell cycle arrest) and HYMAI (imprinted in hydatidiform mole) have been identified as potential candidates. Three genetic mechanisms have been shown to result in TND, paternal uniparental isodisomy of chromosome 6, paternally inherited duplication of 6q24, and a methylation defect at a CpG island overlapping exon 1 of ZAC/HYMAI.
- imprinting
- transient neonatal diabetes
- TNDM
- chromosome 6
Statistics from Altmetric.com
DEFINITION OF TRANSIENT NEONATAL DIABETES
Transient neonatal diabetes is defined as diabetes beginning in the first 6 weeks of life in a term infant with recovery by 18 months of age. A significant proportion of patients go on to develop diabetes in later life and thus the natural history can be split into three phases: phase 1, neonatal diabetes → phase 2, apparent remission → phase 3, relapse of type 2 diabetes.
The incidence in the UK is 1 in 400 000 based on a British Paediatric Association Surveillance Unit (BPASU)) study conducted by Shield et al.1
HISTORICAL FACTS
Reports of neonatal diabetes date back to Ramsey in 1926,2 who reported a male infant with a low birth weight at term who was diagnosed with diabetes at 4 weeks of age, required insulin temporarily, and recovered by 10 weeks. The patient was reassessed at 4 years of age and was reported as healthy. However, the early authors did not distinguish between transient and permanent neonatal diabetes in their reports and it was not until the paper of Hutchinson et al3 in 1962 that this was clarified and they called the condition “Congenital temporary diabetes mellitus”. It was Lawrence in a letter to the BMJ4 who pointed out that it was neonatal and not congenital diabetes and Cornblath and Schwartz5 in 1966 who coined the name “transient neonatal diabetes mellitus”. In 1986, Briggs et al6 drew attention to a female with TND who developed non-insulin dependent diabetes in later life and Shield and Baum7 speculated that TDN might be a risk factor for diabetes in later life. In 1995, Muhlendahl and Herkenhoff8 conducted a long term follow up of published cases and identified 13 infants with TND with later recurrence.
THE SEARCH FOR THE TND GENE
Although there were occasional reports in sibs (Coffey et al9 reported TND in paternal half sibs, and Fergusson et al10 reported diabetes in sibs), as the majority were sporadic a genetic cause was not suspected. The first lead came in 1995 when two children with TND were discovered to have paternal uniparental isodisomy of chromosome 6 (pat UPD(6)).11 The first case was part of a systematic study to identify the incidence of UPD in carriers of supernumerary marker chromosomes (SMCs).12 The patient had few developmental or dysmorphic features and was karyotyped because of intrauterine retardation and a large tongue. She developed diabetes within 24 hours of birth but recovered by 4 months and a diagnosis of TND was made clinically. In published reports at that time, two other cases of pat UPD(6) had been identified.13,14 In both instances the discovery had been made following the development of a rare recessive condition. The first was in a 10 year old child with complete deficiency of complement 4 and no neonatal details were given, and the second developed methyl-malonic acidaemia. However the latter patient, who died at 16 days of age, also had neonatal diabetes. There have now been a further 18 cases reported with paternal UPD(6) and TND15–23 and it has been found to account for 50% of sporadic cases tested.20
In 1996, Temple et al24 discovered that paternal (and not maternal) transmission of a visible duplication of chromosome 6q24 could also lead to TND and this was reproduced in several studies including that of Cave et al,17 who discovered a paternal 6q24 duplication in six of 13 cases with TND. This mechanism accounted for all familial cases. Soon submicroscopic duplications were identified using quantitative PCR.20,25 Overlapping duplications reduced the TND candidate region to 440 kb.25 These observations led to the hypothesis that an imprinted region existed on chromosome 6 and that overexpression of an imprinted and paternally expressed gene caused the condition.
Two imprinted genes were identified within the region, ZAC (zinc finger protein associated with apoptosis and cell cycle arrest) and HYMAI (imprinted in hydatidiform mole),26 shown in fig 1. The region was also found to contain eight CpG islands (CGIs). One CGI was shown to be differentially methylated in normal subjects such that the paternal CGI was unmethylated and the maternal CGI methylated25–27 (named TND CGI). The imprinted locus was also identified using restriction landmark genome scanning by Kamiya et al.27 They showed that ZAC was an imprinted gene in fetal tissues and hypothesised that this was the candidate gene for TND. Varrault et al28 showed that exon 1 of ZAC extended into the imprinted CGI and that unmethylated CG rich sequences within the CGI had promoter activity. This was supported by the work of Arima et al29 who reported that methylation of these sequences silenced promoter activity.
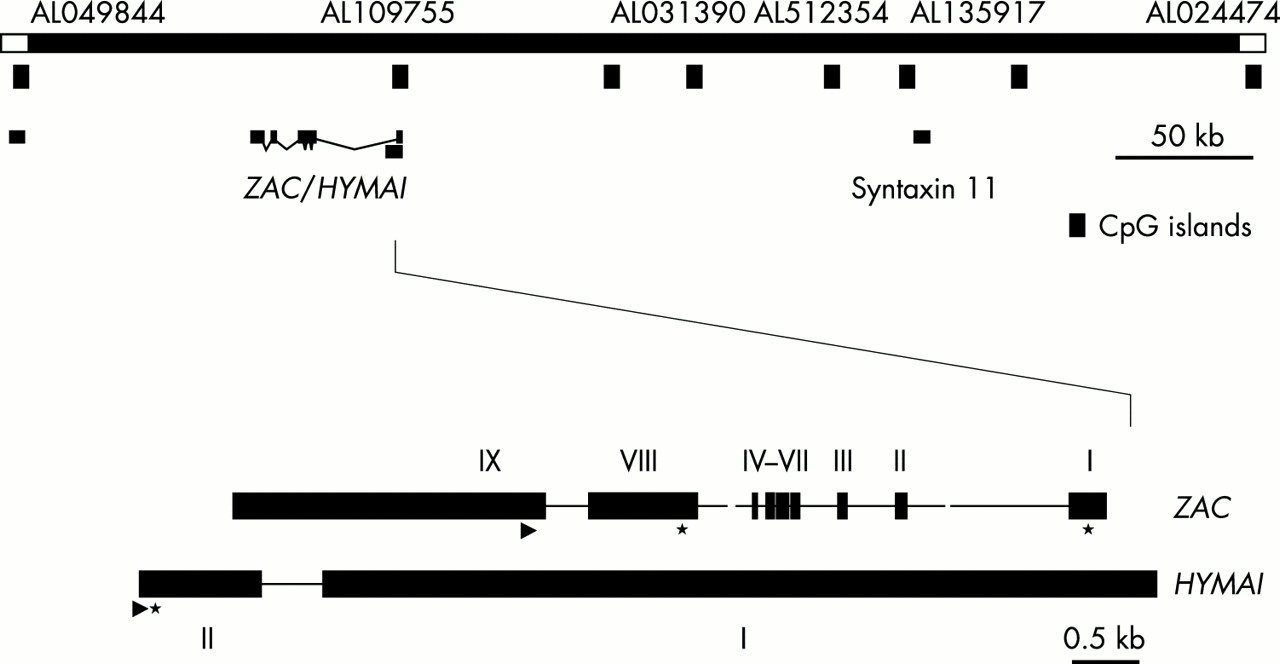
{kind=link}
A map to show the minimal TND critical region defined by the smallest region of overlap in duplication cases.25 The darker hatched CGI overlapping ZAC and HYMAI is differentially methylated (methylated only on the maternal allele). The other CGIs are unmethylated. The sites of the three genes known within the region are marked. The gene structures of ZAC and HYMAI are included but are not to scale. Exon numbering of ZAC is taken from Varrault et al.28
Overexpression of ZAC might cause TNDM through its function as a regulator of cell cycle arrest and apoptosis, altering the absolute number and efficiency of beta islet cells during a critical time of pancreatic development in utero.30 A reduced absolute number of islets might be sufficient to maintain normoglycaemia during the remission phase of the disease but manifest as diabetes at times of metabolic stress, such as intercurrent illness or adolescence. Alternatively we know that ZAC also regulates the expression of the PACAP (pituitary adenylate cyclase activating polypeptide) receptor 1, PACAP being a very potent insulin secretagogue that acts through the PACAP1 receptor in the pancreas.31 It is possible that ZAC overexpression alters normal islet cell responsiveness to PACAP in some as yet unknown manner. HYMAI sequence overlaps ZAC and is also imprinted.26 However, it has no open reading frame and its function is not clear. It remains, however, a potential candidate for TND as to date no mutations have been reported in either gene. Recently, islet cells from a transgenic mouse overexpressing the TNDM locus have shown an inadequate insulin secretory response to glucose stimulation confirming the importance of the locus to normal pancreatic physiology (G Kelsey, personal communication).
In 2000, Gardner et al25 showed that in a number of TND cases the TND CGI was completely unmethylated on both alleles in the absence of uniparental disomy. They termed this a “methylation defect”. Mackay et al31 later showed aberrant expression of both ZAC and HYMAI in a TND patient with a methylation defect. They showed a relaxation of the normal monoallelic expression of both genes in skin fibroblasts of the patient and this provides strong support that the presence of two unmethylated alleles at this locus is associated with inappropriate gene expression.
Therefore, three interrelated genetic mechanisms have now been described in TND: paternal UPD(6), paternal duplication of 6q24, and a methylation defect within the TND CGI. All can be predicted to result in overexpression of a paternally expressed gene with provisional evidence that this is the case for both ZAC and HYMAI.
In a cohort of 30 patients with TND reported by Temple et al20 in 2000, 23 were sporadic cases and seven were familial from two families. One of the three mechanisms was identified as the cause of TND in 24/30 cases studied (80%). Of the sporadic cases 11/23 (≈50%) had pat UPD(6), 4/23 (≈20%) had a paternal duplication of 6q24, and 2/24 had a methylation defect (8%).
CLINICAL FINDINGS: THE 6q24 PHENOTYPE
The genetic classification of TND cases has allowed the 6q24 phenotype to be differentiated from other causes of neonatal diabetes.
Phase 1: neonatal diabetes
The main findings in the neonatal period are growth retardation at term, an onset of diabetes within the first week of life, and recovery within three months. Based on the study of Temple et al20 in 2000, the average birth weight was 1930 g at 39 weeks’ gestation. The median age at presentation was 3 days and the average age at presentation was 7 days. The median length of time on insulin was 12 weeks (average 111 days). Glycosuria is often discovered incidentally during investigation for dehydration and weight loss and some of the differences in age of presentation in published reports are likely to be because of delay in diagnosis.
Another feature noted in the neonatal period by many authors is macroglossia. This was found in 1/3 of cases in the series of Temple et al20 and was not related to any specific genetic mechanism. An umbilical hernia is also occasionally described.20 These findings remain unexplained although it is of interest that the tongue and umbilicus are also abnormal in Beckwith-Wiedemann syndrome, which is another condition resulting from aberrant imprinting mechanisms.
The systematic investigation of neonatal diabetes in patients with TND is difficult, as the condition is so rare. However, in those where further studies have been performed, insulin levels were low or undetectable at presentation but ketonuria was usually absent.1,8,17,20 There was no association with HLA antigens common in type 1 diabetes1 and there was no evidence that TND was the result of autoimmunity. Islet cell antibodies were negative.20
Phase 2: apparent remission
This phase is associated with normal growth and development in the majority.20 There is evidence that patients may develop hyperglycaemia during periods of intermittent illness (personal observation). Few studies have been performed on patients during this remission phase and none has yielded consistent results. Seven patients were studied with a standard intravenous glucose tolerance test (IVGTT) by Shield et al (personal communication) during apparent remission. Four cases had low insulin secretion but in three it was normal.
Phase 3: relapse of diabetes
Relapse has been reported in approximately 50-60%20 of TND cases although no long term follow up of cases diagnosed at birth has yet been reported so this figure is likely to be an over-ascertainment. Of 15 relapsed cases studied in the UK, the earliest age of relapse was 4 years and the average age was 14 years (personal observation). Treatment in this group varied between diet alone (4/15), oral hypoglycaemic agents (2/15), and insulin (9/15). The variability of treatment modalities has been suggested to imply that this condition is more akin to type 2 diabetes.1 It should be noted that often the exogenous insulin requirement is lower than that usually seen in type 1 diabetes, may be intermittent, and there is some evidence to support insulin resistance in two cases studied by Shield et al.32
BROADENING THE PHENOTYPE
In 2000, Temple et al20 reported two cases that suggested that the 6q24 phenotype might be broader than that described above. Two relatives with identical paternal 6q24 duplications to the probands were identified who presented first in adulthood with type 2 diabetes and no early history. One was the paternal aunt of the sibs first reported by Fergusson and Milner,10 who developed gestational diabetes and was treated by diet alone, and the other was a male sib in an unrelated family who developed type 2 diabetes in his teens. This observation led to studies to discover the incidence of chromosome 6 anomalies (as seen in TND) in cohorts of patients with other types of diabetes. These have all been negative and included the only published study by Shield et al,33 who looked at a cohort of MODY cases where known MODY gene mutations had been previously excluded. A cohort of 65 type 2 diabetics has also been studied but with similar negative results (D J G Mackay, personal communication).
It must be concluded therefore that the clinical presentation of an imprinted 6q24 anomaly may not invariably include a neonatal presentation, but that this is not a frequent cause of commoner types of adult diabetes. However, in 2001, Lindsay et al34 showed weak linkage of diabetes in Pima Indians to paternally derived alleles at 6q24 (lodAF=0.9) and this locus may yet be shown to play a role in more common types of diabetes perhaps through common polymorphic variants.
GENETIC COUNSELLING
No phenotypic differences have been found between patients with UPD(6), duplication of 6q, and a methylation defect. The classical natural history of the condition can therefore be used for counselling purposes for all groups of patients with the added proviso that occasionally a child identified, perhaps in utero, may not necessarily develop diabetes in the neonatal period but is at risk of developing it in the long term.
Cases with UPD(6) are predicted to be sporadic with a low recurrence risk to both sibs and offspring. A duplication of 6q24 has been found in all familial cases to date. If the duplication is passed on by a female the offspring risk of developing TND is low but TND may occur in subsequent generations. Males with a duplication will have a 50% risk of passing it on and having an affected child; however, as discussed above it is not inevitable that the child will present in the neonatal period. The sib and offspring risk for the methylation cases are not known. Of seven cases with a methylation defect all have been sporadic (personal observation). However, it is possible that a maternal mutation/deletion (not identified by the technique used to show the abnormal methylation pattern) may be present and could theoretically result in recurrence risks for sibs and offspring as high as 50%, but this is speculative at present.
DIFFERENTIAL DIAGNOSIS
When a patient presents with diabetes in the neonatal period, there are a number of other conditions that must be considered. Cave et al17 estimated that TND accounted for only 50-60% of cases of diabetes presenting in the neonatal period. In the absence of a demonstrable chromosome 6 abnormality it is not possible to differentiate TND from permanent neonatal diabetes mellitus (PNDM) in the neonatal period. PNDM is usually diagnosed by the natural history and is defined as diabetes developing in the neonatal period that never goes into remission. There are likely to be many causes. Stoffers et al35 described the first case to have a specific gene mutation in 1997, when they identified a child with pancreatic agenesis homozygous for a single nucleotide deletion in the IPF-1 gene encoding the homeodomain protein IPF-1. This protein is a critical regulator of insulin gene function in the islet cell. Imaging of the pancreas may help to differentiate such cases.
Wolcott-Rallison syndrome36 is an autosomal recessive disorder characterised by infancy onset (often within the neonatal period) diabetes and spondyloepiphyseal dysplasia. The skeletal findings may develop later, however, and this condition is the result of mutations in EIF2AK3 at 2p12.37 This gene is highly expressed in islet cells acting as a regulator of protein synthesis.
X linked immunodysregulation, polyendocrinopathy, and enteropathy (IPEX)38 is a disorder of autoimmunity that occurs in males presenting with neonatal diabetes, immune dysregulation, intractable diarrhoea, eczematous or psoriataform skin rash, and very raised levels of IgE. It is usually lethal in the first months or years of life from overwhelming sepsis.
X linked phosphoribosyl-ATP pyrophosphatase hyper-activity39 causes diabetes early in life and glucose intolerance may persist throughout life. However, it is associated with major problems including mental retardation, ataxia, and a progressive axonal neuropathy.
Glucokinase deficiency (MODY2) is caused by mutations in the glucokinase gene40 and usually leads to mild hyperglycaemia in affected subjects. However, within two families (one Norwegian, one Italian) two infants presented with classical PNDM who were homozygous for missense mutations within the glucokinase gene rendering them completely deficient in glycolytic activity.41 A further syndrome is an autosomal recessive condition of neonatal diabetes and cerebella hypoplasia.42 The infants all died within a few months of birth and the genetic basis is unknown.
CONCLUSION
TND is a rare but distinct cause of neonatal diabetes with a good prognosis compared to many other causes of childhood diabetes. It is the result of an imprinted gene(s) at 6q24 and the genetic cause can be identified in 80% of cases.20 This locus may play a role in other more common types of diabetes.
Acknowledgments
Dr I K Temple and Dr J P H Shield are part of the European TND network. We would like to thank Dr D J G Mackay for fig 1 and for help with the manuscript.