Article Text

Abstract
Background: Silver–Russell syndrome (SRS) is a clinically and genetically heterogeneous condition characterised by severe intrauterine and postnatal growth retardation. Loss of DNA methylation at the telomeric imprinting control region 1 (ICR1) on 11p15 is an important cause of SRS.
Methods: We studied the methylation pattern at the H19-IGF2 locus in 201 patients with suspected SRS. In an attempt to categorise the patients into different subgroups, we developed a simple clinical scoring system with respect to readily and unambiguously assessable clinical features. In a second step, the relationship between clinical score and epigenetic status was analysed.
Results and conclusions: The scoring system emerged as a powerful tool for identifying those patients with both a definite SRS phenotype and carrying an epimutation at 11p15. 53% of the 201 patients initially enrolled fulfilled the criteria for SRS and about 40% of them exhibited an epimutation at the H19-IGF2 locus. Methylation defects were restricted to patients who fulfilled the diagnostic criteria for SRS. Patients carrying epimutations had a more severe phenotype than either the SRS patients with mUPD7 or the idiopathic SRS patients. The majority of patients with methylation abnormalities showed hypomethylation at both the H19 and IGF2 genes. However, we also identified SRS patients where hypomethylation was restricted to either the H19 or the IGF2 gene. Interestingly, we detected epimutations in siblings of normal parents, most likely reflecting germ cell mosaicism in the fathers. In one family, we identified an epimutation in an affected father and his likewise affected daughter.
Statistics from Altmetric.com
Key points
Loss of DNA methylation at the imprinted IGF2-H19 locus is an important cause of SRS.
SRS patients carrying epimutations have a more severe phenotype than patients with mUPD7 or idiopathic SRS patients.
The majority of patients with methylation abnormalities show hypomethylation at both the H19 and IGF2 genes.
A subset of SRS patients carry epimutations restricted to either the H19 or IGF2 gene.
This study reports, for the first time, familial cases of hypomethylation at the 11p15 locus.
Silver–Russell syndrome (SRS, MIM 180860) is a clinically heterogeneous condition characterised by severe intrauterine growth retardation, poor postnatal catch up growth, craniofacial features such as a triangular shaped face and a broad forehead, body asymmetry and a variety of minor malformations. The phenotypic expression changes during childhood and adolescence and the facial features and asymmetry usually become much more subtle with age. Although different groups have established diagnostic criteria for SRS, diagnosis often remains doubtful and children with intrauterine growth retardation of various aetiologies are spuriously diagnosed with SRS.1 2 This has caused confusion with regard to the true incidence and natural history of SRS.
It has been recognised for a long time that SRS, which mostly occurs sporadically, is very likely a genetically heterogeneous condition with a complex genetic background.3 However, until recently a genetic defect could only be identified in a small subset of patients with SRS: maternal UPD7 in about 5–10% of SRS cases and a chromosomal rearrangement such as maternal dup(11p15) and dup(7p11.3-p13) or segmental mUPD(7q31-qter) in <1%.4–9 This situation changed in 2005 when hypomethylation of the differentially methylated IGF2/H19 imprinting centre region (ICR1) at 11p15 was reported in a small number of SRS patients.10 This finding was soon confirmed in larger cohorts of SRS patients and it turned out that partial or complete loss of DNA methylation at the telomeric ICR1 on 11p15 represents a major cause for SRS.11–14 Interestingly, opposite epimutations to the ones observed in SRS, namely hypermethylation of IGF2/H19, are observed in about 5–10% of patients with the Beckwith–Wiedemann syndrome (BWS), an overgrowth syndrome representing in many respects the countertype to SRS.15
The 11p15 region comprises two imprinted domains important for the control of fetal and postnatal growth, ICR1 and ICR2. ICR1 coordinates the expression of two oppositely imprinted genes, H19 and IGF2. H19 encodes a non-translated RNA and is expressed exclusively from the maternal allele.16 Hypomethylation of H19 in SRS is proposed to lead to biallelic expression of H19, down regulation of IGF2 and hence growth restriction. How the 11p15 epimutation causes the specific SRS phenotype is not completely understood at the moment. Up to now about 100 patients—mainly children—with 11p15 epimutations have been reported in the literature. However, the phenotypic spectrum of epimutation carriers and their long term perspective still remain vague.
The present study aimed to determine more precisely the clinical phenotype(s) associated with epimutations at the 11p15 region. We wanted to determine whether the patients with methylation defects might comprise a distinct group from idiopathic SRS patients and the ones with mUPD7. As a first step we developed a simple clinical scoring system with regard to the severity of the SRS phenotype. Then, in a second step, we analysed the relationship between clinical score and epigenetic status.
PATIENTS AND METHODS
Study population
The study population consisted of 201 individuals with suspected SRS (table 1). The patients were followed in one of the genetic clinics participating in this study (Zurich, Warsaw, Minsk, Istanbul) between 1990 and 2007, with the exception of 15 individuals whose blood samples were sent to Zurich for genetic analysis. Informed consent for analysis was obtained from all individuals included in the study—that is, adults (patients and parents) and parents of underage patients. The study was performed in accordance with national ethics guidelines.
Clinical scoring system
Each patient was physically examined by a geneticist in one of the participating centres. The mean age at physical examination, usually coinciding with the blood sampling, was 6.5 years, with a range from 1–42 years of age. A form with information concerning birth measurements, perinatal adaptation, postnatal course, dysmorphic signs, feeding behaviour, intellectual development and family history was completed by the examining physician. Facial features and body asymmetry were assessed from hospital charts or childhood photographs in those patients who had already reached adulthood (about 5% of the study sample). The clinical data of all the patients were entered into a database. Upon gathering the entire body of clinical information of the cohort we decided to omit information on bone age, target height, feeding difficulties, hypoglycaemia, sweating and teeth abnormalities; although these symptoms represent important diagnostic features in SRS, they were often reported imprecisely or were missing. We therefore decided to retain in the database only those clinical features of the patients that were readily and unambiguously—and therefore reliably—assessable. Using the clinical criteria for SRS defined by Price et al in 1999 as its basis, we established a refined “SRS severity” scoring system. Each patient was scored for the five domains: (1) parameters at birth; (2) postnatal course; (3) asymmetry; (4) facial features; (5) other features (table 2). For four of the five domains three features were scored and 0–3 points were given, with 0 depicting that none of the three features was present, 1 that one was present, etc. Asymmetry was assessed in a different way; since this is a very important feature in SRS but difficult to quantify we decided to score this feature simply as present (3 points) or missing (0 points). According to this procedure a minimum total score of 0 points and a maximum total score of 15 points was assigned.
Scoring of all the patients (on the basis of the information collected in the database) was performed by one of the authors who was not involved in examination of the patients and who was blind with respect to the findings of the molecular analysis. When scoring of the cohort was completed, we carefully compared the clinical presentation of those patients who displayed scores in the middle range (between 6–10 points). It was arbitrarily defined that patients with a score ⩾8 points were classified as SRS patients whereas those with lower scores were not (non-SRS patients).
Statistical analysis
The characteristics of the population were described as percentage for qualitative traits or as median, mean and SD for quantitative traits (clinical scoring). The relationship between the severity score and the epigenetic status (H19/IGF2 hypomethylation group, H19 hypomethylation group, mUPD7 group, idiopathic SRS group) was assessed by one way analysis of variance (ANOVA). Values of p<0.05 were considered to indicate significance.
Methylation analysis for the 11p15 region
DNA was extracted from peripheral blood samples of all individuals: control individuals, patients, and at times parents and siblings. One hundred age matched healthy individuals served as controls.
The methylation status at H19 and IGF2 was investigated by multiplex ligation dependent probe amplification (MLPA) (ME030, MRC, Holland, version 03, 3-1-2007) according to the protocol supplied by the manufacturer. Briefly, approximately 100 ng DNA of each individual were first denatured and hybridised overnight with the mixture of probes (supplied with the kit), and then, after splitting the sample in two, treated either with ligase alone or with ligase and HhaI. Polymerase chain reaction (PCR) reactions were then performed with the reagents and primers supplied in the kit. The PCR products were separated by capillary electrophoresis (ABI310). The relative ratio of methylation at H19 and IGF2 were calculated as follows: the areas of the peaks of interest were divided by the average peak area of the closest control peaks. For each peak, the relative area of the sample treated with HhaI was divided by the value obtained for the undigested sample. These ratios were normalised by dividing by the average ratios obtained for control individuals. Mean (SD) values of the 100 control individuals are depicted in the caption to fig 1. The results obtained for the probes 6266-L5772 H19DMR (301 bp) and 7175-L6784 IGF2 (AY375532.1, 9046-9047) DMR0 (211 bp) were in agreement with previous methylation analysis by MSP/DHPLC (data not shown) and were not subject to high variability due to the presence of polymorphisms in the target sequences.
{kind=link}
Analysis for mUPD7
The possible occurrence of maternal UPD7 of the two critical regions on chromosome 7 (7p11.2-p13 and 7q32-34) was investigated before this study (data not shown). Analysis was performed as described in one of our earlier reports by genotyping DNA samples from the patient, mother, and father with 13 chromosome-7-specific polymorphic microsatellite markers.19 At least one and preferably two informative markers per region were considered necessary to exclude UPD.
Analysis of familial cases
In the three families with either two affected siblings or vertical transmission (father and daughter affected) we sequenced the CTCF binding sites in the DMR 5′ of the H19 gene. Primer sequences were as reported previously.12 To document the methylation status at another paternally imprinted locus the DMR of the DLK1-GTL2 locus on chromosome 14q32 was analysed, as described previously.20
RESULTS
Clinical scoring
The characteristics of the study population are summarised in tables 1 and 3. According to the scoring system described above, 106 individuals had scores of ⩾8 points and were classified as SRS, and this constituted 63% of all the patients scored. A diagnosis of SRS was accordingly excluded in 62 individuals (37%). These non-SRS individuals were primarily patients with prenatal growth retardation. No definitive diagnosis could be established in these patients.
Chromosomal analysis (data not shown)
One individual with a female phenotype turned out to have a male karyotype in blood lymphocytes (46,XY). This patient also carried an epimutation at 11p15 and will be reported on in detail in a separate publication. The remainder of the individuals had normal karyotypes.
Molecular evaluation
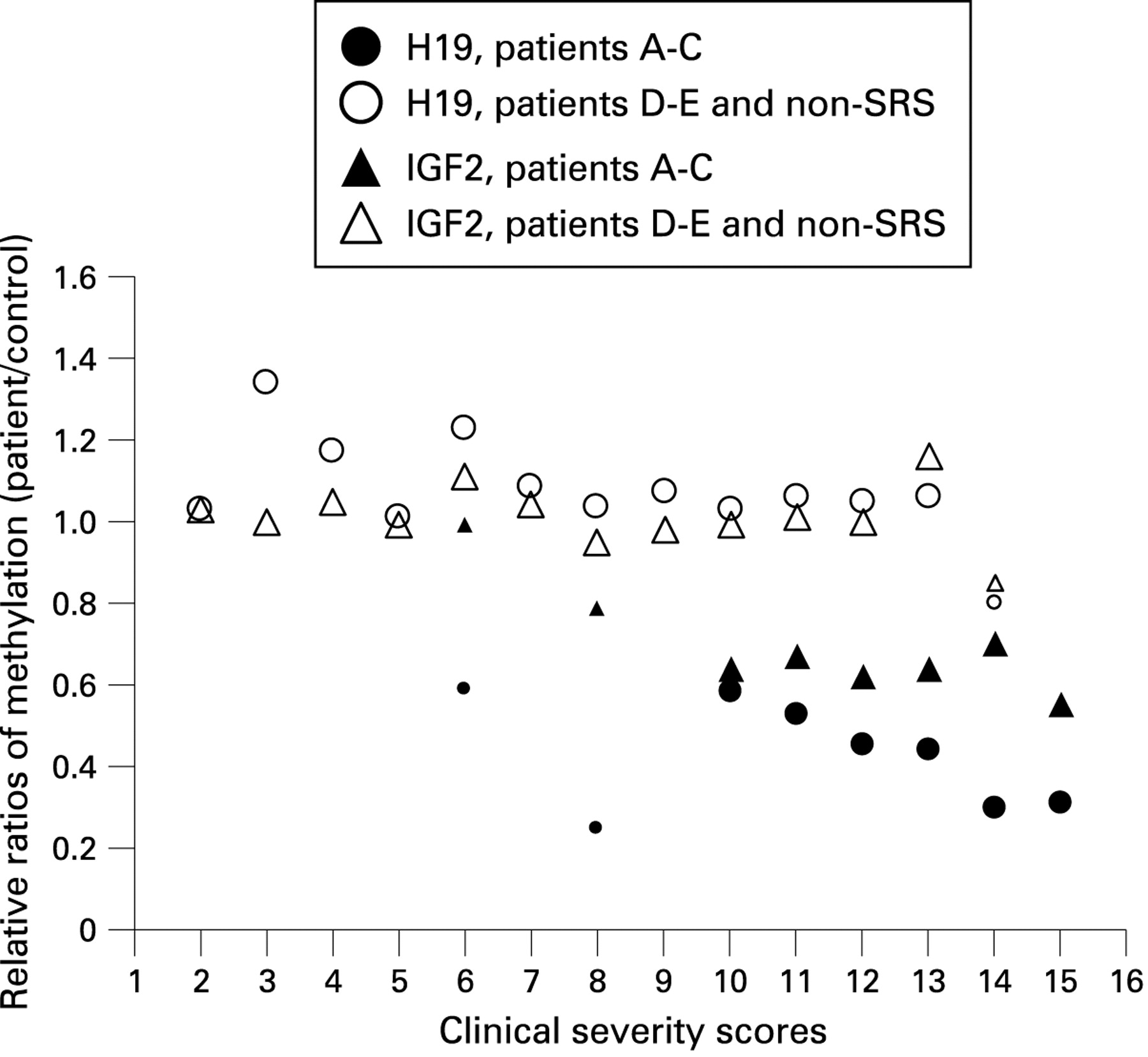
Epimutations at 11p15 are specific for SRS and are not a cause of non-specific pre- or postnatal growth retardation (tables 3 and 4, fig 1)
In 46 patients, hypomethylation at the ICR1 on 11p15 was detected. The clinical dataset was incomplete in four of these and they were therefore excluded. From the 42 remaining patients, 41 had a score of ⩾8 points and were therefore classified as SRS. From our cohort of 200 patients, we could therefore demonstrate that epimutations at 11p15 were specific to patients with classical SRS and were not present in patients, primarily children, either with isolated growth retardation, body asymmetry or minor anomalies similar to those occurring in SRS. There was only one exception to this, a female patient in mid childhood who had a score of 6 and therefore did not fulfil the criteria for SRS. This girl actually exhibited a partial hypomethylation specific to H19 (not IGF2). She showed borderline prenatal and pronounced postnatal growth retardation, no facial dysmorphisms, mild brachyphalangy, and no asymmetry. Unfortunately it was not possible to organise a follow-up visit for this girl to verify her clinical profile.
Epimutations at 11p15 are restricted to the telomeric gene cluster and are not always congruent for the H19 and IGF2 gene (table 4, fig 1)
The MLPA technique applied allowed us to analyse the telomeric and the centromeric gene cluster on 11p15 simultaneously. In our cohort no methylation defects were detected at the centromeric cluster where CDKN1C and KCNQ1OT1 are located. Differential analysis of the H19 and IGF2 genes in the telomeric cluster revealed that the majority of patients showed hypomethylation of both H19 and IGF2. Interestingly, 11 patients showed selective hypomethylation of H19 and three patients showed selective hypomethylation of IGF2. The IGF2-specific probe showed a broader variation in controls as compared to the H19 probe and these results should therefore be interpreted with some reservation.
Epigenetic–phenotype correlation (table 4, fig 1)
Patients carrying epimutations had higher scores then the ones with mUPD7 or the idiopathic SRS patients, indicating that hypomethylation at 11p15 was associated with a severe SRS phenotype. Median severity scores, means and SDs are depicted in table 4. The differences of the severity scores were highly significant when comparing the group with combined hypomethylation of H19 and IGF2 with the mUPD7 patients (p<0.001) or the idiopathic SRS patients (p<0.001) and still significant when comparing the group with hypomethylation of H19 to the mUPD7 and idiopathic SRS group (p<0.01). The scores of the mUPD7 group were not significantly different when compared to the idiopathic SRS group (p>0.05). Since we identified only three patients with hypomethylation restricted to IGF2, no statistical analysis could be performed for this group.
The methylation index, reflecting the degree of hypomethylation in a given individual, seemed to correlate with the score and therefore with clinical severity, at least for H19—that is, the relationship between hypomethylation of H19 and clinical severity might be quantitative. The small number of patients did not allow statistical analysis with regard to this point.
Our scoring system was not designed to establish a correlation between the presence of individual features and the epigenotype. However, it was obvious that asymmetry was more frequent in those SRS patients with a methylation abnormality when compared to the other groups: 65% (27 of 42) of SRS patients with a methylation defect exhibited body asymmetry, versus 40% (23 of 58) of idiopathic SRS patients, 20% (2 of 10) of patients with mUPD7, and 5% (3 of 62) of non-SRS patients.
Familiar cases with epimutations at 11p15
Two families, each with two children who displayed hypomethylation of H19 and IGF2, were identified. In both families neither of the parents showed clinical signs of SRS, and methylation analysis in both fathers revealed normal results. In the first family a school aged boy and a girl were affected, both of them showing a classical SRS phenotype with asymmetry of the limbs in the boy and an asymmetrical face in the girl. In the second family the affected siblings were both girls. A report on the second family had already been published before epimutations at 11p15 were demonstrated in SRS.21 Both girls were scored at 14 points, reflecting the severe phenotype. In a third family we detected an epimutation at 11p15 in both a clinically affected father in his third decade (displaying a score of 10) and his affected daughter (score of 12 points). None of the familiar cases showed any clinical symptoms in addition to the SRS phenotype.
Sequence analysis of the CTCF binding sites in the DMR between the IGF2 and the H19 genes did not reveal any mutations, and analysis of the paternally imprinted DLK1-GTL2 locus on chromosome 14q32 showed a normal methylation pattern in the three families.
Maternal uniparental disomy (mUPD7) (table 4, fig 1)
Ten individuals displayed maternal uniparental disomy of chromosome 7. Interestingly, only seven out of these 10 individuals fulfilled the diagnostic criteria for SRS according to our scoring system. The seven patients with SRS phenotypes showed significantly lower scores when compared to the ones displaying hypomethylation of 11p15. Comparison with the idiopathic SRS patients did not show a statistically significant difference in the severity scores.
DISCUSSION
This study of 201 individuals with SRS and SRS-like phenotypes confirms the high frequency of methylation abnormalities at the H19/IGF2 locus in SRS: epimutations occurred in about 40% of patients with SRS. In total, we identified more than 40 individuals with partial hypomethylation at the ICR1 on 11p15, which is the largest cohort of SRS patients with epimutations reported in the literature so far. Methylation defects were, with one exception, restricted to patients who fulfilled the diagnostic criteria for SRS.
Establishing the diagnosis of SRS is not always easy and there is no general consensus for diagnostic criteria. This has added confusion to the question of frequency of 11p15 epimutations in SRS. While the French group, which was first to describe methylation abnormalities on 11p15 in SRS, detected epimutations in 64% of a large series of SRS patients, other groups identified this abnormality in their cohorts at lower frequencies of 30–40%.22 Differences in detection rates might be explained to some extent by the molecular techniques used: it was recently shown that MLPA is more sensitive than Southern blot analysis for assessment of the methylation status at 11p15.23 Much more important, however, are the criteria used for establishing the SRS diagnosis in a given individual. The frequency of 40% epimutation carriers in our cohort therefore reflects the fact that we included patients with a broad clinical spectrum from borderline to classical SRS phenotypes.
To address the question of clinical heterogeneity and stringency of diagnostic criteria in SRS we developed a very simple scoring system which permits calculating an SRS severity score ranging from 0–15 points. The advantage of this scoring system is that it allows the determination of the phenotype on a quantitative basis. It emerged that patients with hypomethylation had significantly higher scores and therefore more severe phenotypes when compared to the idiopathic SRS patients where no genetic abnormality was identified or the patients with mUPD7: 95% of the patients with a methylation abnormality had scores between 10–15 points, whereas only 46% of the idiopathic SRS patient were within this range. This has important implications for the diagnostic strategy in an individual with SRS. The chance of detecting a methylation abnormality in a child with SRS and a low score of 8 or 9 points is very small and analysis for mUPD7 would be recommended first. On the contrary, methylation analysis should be the first diagnostic step in those patients with high scores between 10–15 points, especially in individuals with asymmetry. One might speculate that the milder phenotype of the SRS patients in whom we could not identify a genetic abnormality might carry a methylation abnormality on 11p15 which escaped analysis in blood samples. It is well known from BWS that methylation defects at the11p15 locus mostly occur as somatic mosaics and that the detection rate depends on the tissue analysed. It would be interesting, therefore, to test other tissues—for example, fibroblasts—for methylation abnormalities in those “blood negative” patients.
IGF2 is a paternally expressed fetal growth factor and H19 a maternally expressed non-coding RNA for which the function is still not very well understood. Imprinted expression of these two genes is regulated by the action of the H19 DMR. It largely remains unknown how biallelic silencing of IGF2 and activation of H19 translates into the specific SRS phenotype. It has been shown that hypomethylation of H19 in SRS patients is associated with decreased levels of IGF2 mRNA in fibroblasts, but IGF2 serum values seem to be mostly normal in epimutation carriers.10 13 24 This observation is not surprising given the development and tissue dependent expression pattern of this growth factor, with monoallelic expression from the paternal allele in fetal life and biallelic expression in postnatal life.25
We observed selective hypomethylation for either H19 or IGF2 in 13 patients, indicating that hypomethylation of IGF2 alone seems to be sufficient to cause classical SRS. The phenotype of those patients with selective hypomethylation at one locus was generally milder than in the patients displaying hypomethylation of H19 and IGF2. However, given the small number of patients (11 for selective H19 and two for IGF2 demethylation) this observation needs to be confirmed. To date there is limited knowledge on the methylation status of IGF2 in SRS and it is not know to what extent the proposed silencing contributes to the SRS phenotype. A recent paper on methylation changes at the IGF2/H19 locus in SRS, BWS and Wilm’s tumour reports on similar methylation abnormalities at H19 and IGF2 in the congenital growth disorders, but different methylation profiles in tumours.26 The authors propose different mechanisms of imprinting loss in neoplastic cells and growth disorders. Along these lines it remains open whether loss of imprinting in those SRS patients with combined H19/IGF2 hypomethylation is caused by a different mechanism than in the cases with selective hypomethylation of H19 or IGF2.
In this study we describe for the first time familial cases of hypomethylation at the 11p15 locus. This finding has important implications for both genetic counselling and determination of the origin of the epimutations. The setting of epigenetic marks in germ cells has been studied in detail in mouse and to a lesser extend also in humans.27 Methylation marks at imprinted loci are thought to be erased in primordial germ cells and then reacquired during spermatogenesis and oogenesis. In the majority of epimutation carriers identified in this study the loss of the paternal methylation mark may have resulted from a deficient acquisition of methylation during spermatogenesis or from lack of maintenance of the methylation mark on the paternal allele after fertilisation. The two families for whom we identified epimutations in siblings most likely represent germ cell mosaicism of an incorrect methylation mark at the ICR1 during spermatogenesis in the fathers.
The third family where the epimutation was transmitted from an affected father to his daughter is the first case of vertical transmission through the male germ line described in the literature. The underlying mechanism is difficult to explain in this family. Obviously the affected father displays a missing methylation mark at the H19/IGF2 DMD in somatic and germ cells. Reprogramming of the methylation pattern in his primordial germ cells did not lead to a correction (setting of a methylation mark), but the “mistake” was continued when resetting of the methylation pattern at imprinted loci occurred during spermatogenesis. How this mistake was “memorised” in the germ cells of the father remains to be explained. Father and daughter showed a classical SRS phenotype and did not have additional symptoms indicative of a different aetiology compared to the sporadic cases. Both displayed partial hypomethylation at the H19/IGF2 DMD and analysis of another paternally imprinted locus at 14q (DLK1-GTL2) showed a normal methylation pattern. Therefore, although the clinical picture and the molecular findings do not point in this direction, it cannot be excluded that father and daughter carry a mutation in one of the DNA methyltransferase genes or another gene involved in DNA methylation.
It is interesting to note that the four children of the two families with suspected germ cell mosaicism in the fathers displayed hemihypoplasia. It has been hypothesised that asymmetry in BWS and SRS is caused by mosaic distribution of post-zygotically acquired epimutations.28 However, the observation of asymmetry in the offspring of men with suspected germ cell mosaicism for epimutations at the H19/IGF2 locus renders it rather unlikely that asymmetry can be explained by mosaic distribution of the epimutation. In those children the epimutation most likely did not emerge post-zygotically and the asymmetry has therefore to be explained by another still unknown mechanism.
In summary, our study has important implications for diagnosis and genetic counselling in SRS. First, the clinical scoring system we developed is a simple but powerful tool to direct the diagnostic efforts rapidly towards methylation analysis in those patients with a severity score in the upper range. Second, we demonstrate that hypomethylation of H19 or IGF2 alone appears to be sufficient to cause SRS. Third, and most importantly, the observation of germ cell mosaicism and vertical transmission from an affected father to his child demonstrates that there is a recurrence risk for epimutations in SRS.
Acknowledgments
We thank the patients and their families, the referring physicians and our former and present colleagues from the IMG who supported this study. Special thanks go to Dr D Kotzot who is now affiliated with the University of Innsbruck.
REFERENCES
Footnotes
Competing interests: None.
Funding: The study was supported in part by the Swiss National Science Foundation (Grant number: 3200B0-105536, to AB).
Patient consent: Not required