Article Text

Statistics from Altmetric.com
- OI, osteogenesis imperfecta
- NAI, non-accidental injury
- EDS, Ehlers-Danlos syndrome
- CPS, Child Protective Services
- DI, dentinogenesis imperfecta
Non-accidental injury (NAI) is a major public health concern that affects an estimated 800 000 children each year (National Child Abuse and Neglect Data System, 1999, http://www.acf.dhhs.gov/programs/cb/publications), and involves their parents and family members, physicians, nurses, social workers, and others who participate in the care and evaluation of these children. Health care providers in the United States are compelled by federal law (Child Abuse Prevention and Treatment Act, 1974) to report suspected cases of child abuse. Although abuse is common, it may be difficult to discern the true cause of an injury and it is estimated that approximately 7% of children who have signs suggestive of abuse actually have an underlying medical condition that explains their injuries.1 Some of the more common genetic conditions that have clinical features which share features of NAI include osteogenesis imperfecta (OI),2 several types of Ehlers-Danlos syndrome (EDS),3,4 and glutaric acidaemia type I.5 In its milder forms, OI may present simply as unexplained bone fractures in childhood.2
When children present to the accident and emergency department or clinic with unexplained fractures, they and their parents are usually referred to Child Protective Services (CPS) for at least a preliminary evaluation. Depending on the results of this evaluation, infants and parents can be separated for anywhere from a few minutes to weeks or months, until the investigation is terminated. If the child who is separated from the family is shown subsequently to have OI, for example, the separation can have devastating effects on the family and their relations to medical and other health care or social service personnel. Conversely, parents who have abused their child may claim that the child has OI.6,7 In some instances, the return of previously abused children to the family can lead to further injury and even death.
OI is a genetic disorder that occurs in approximately 1:10 000 births.8–10 NAI with fractures occurs in approximately 24:10 000 children (National Child Abuse and Neglect Data System, 1999) in the birth to 3 year age range, so that a child is about 24 times more likely to have broken bones secondary to abuse than to OI. The diagnosis of OI depends on obtaining a careful family history, review of the x rays, and physical examination in which characteristic features (for example, blue sclerae, short stature, fractures, bone deformity, dentinogenesis imperfecta, and hearing loss, many of which have age dependent penetrance11) are sought or identified. To recognise children with OI among those with fractures requires that physicians are familiar with OI as an entity and know how to make the clinical diagnosis. Often there is no family history of OI, and radiographic studies may be equivocal.2,8,12 The clinical differentiation of OI and NAI may be difficult. Fractures in various stages of healing can be seen in both, there may be a family history of fracture because of abuse, pale blue sclerae (one of the commonest features mentioned in examinations) are normal in young children,13,14 and some features of OI, such as DI and hearing loss, are not apparent in infancy.
The biochemical bases of most forms of OI were identified in the 1970s and shown to alter the amount or structure of type I collagen, the major protein of bone, synthesised.15 The first mutations were identified in the early 1980s16–19 and since then a few hundred mutations in the two type I collagen genes, COL1A1 and COL1A2, have been described2 (Database of Human Type I and Type III Collagen Mutations; http://www.le.ac.uk/genetics/collagen/). These studies provided the basis for the biochemical20 and molecular genetic testing21 that is now available to assist in the diagnosis of OI. These studies take two forms. In the first, skin fibroblasts are grown and the amount and structure of the type I procollagen molecules synthesised by the cells is measured. This approach identifies almost 90% of subjects known to have OI.20 In the second approach, the coding regions of the two genes (COL1A1 and COL1A2) that encode the chains of type I procollagen are screened for pathogenic sequence alterations, a strategy that also identifies about 90% of subjects known to have OI.21
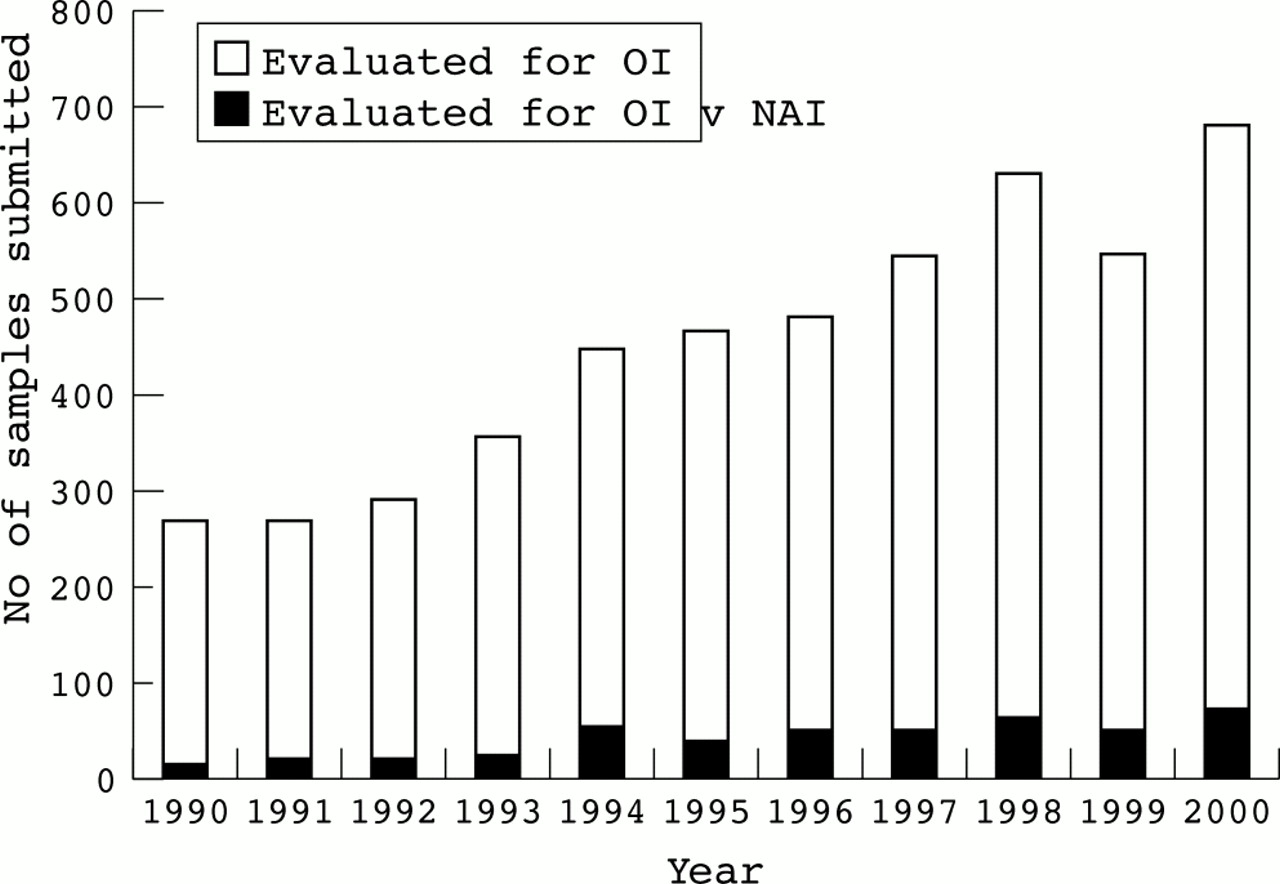
Currently, we study approximately 600 specimens each year from adults and children in whom the diagnosis of OI is being considered. About 10% of these tests are ordered in instances in which NAI is suspected, but OI is considered or cannot be excluded (fig 1). To determine the efficiency with which these studies identified children with OI among those in whom abuse was considered, we reviewed the clinical information provided for 262 samples in the context of the results of our biochemical studies. Cells from 11 of these infants had abnormalities consistent with OI and in 11 others the diagnosis could not be excluded. These findings confirm that these analyses have a place in the evaluation of children in whom the diagnosis of OI cannot be excluded on the basis of physical examination alone. Given the sensitivity of collagen screening and the relative frequencies of OI and NAI, it appears that only a small proportion of children with OI will be missed by this approach.
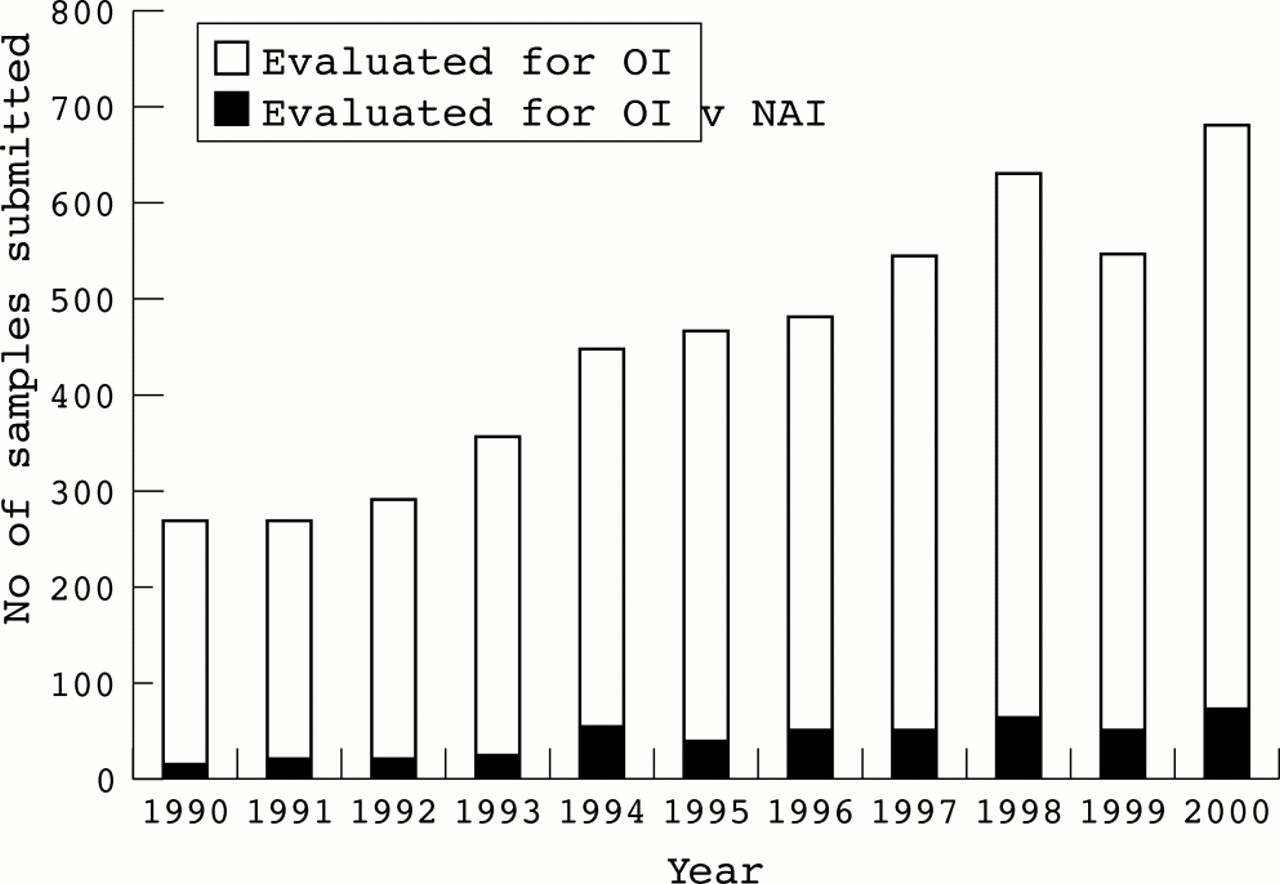
Number of samples submitted for biochemical tests for osteogenesis imperfecta to the Collagen Diagnostic Laboratory at the University of Washington, 1990-2000. The number of samples submitted each year is indicated by the height of the white bar and the black bar indicates those submitted for an evaluation when abuse was also considered.
SUBJECTS AND METHODS
Clinical evaluation and data collection
We reviewed the clinical data provided to us about all subjects with a diagnosis of OI, possible OI, or fractures of unknown origin whose cells were sent to us to examine collagen synthesis and structure from 1 January 1998 to 28 February 2001 (n=1857). We identified those in whom abuse (non-accidental injury, NAI) was being considered to explain fractures; 263 such children were identified, all but seven under the age of 3 years. The mean age of the infants studied was 9.4 months with a median age of 6.0 months (fig 2). In one instance cells submitted did not grow. Studies of collagen synthesis and structure were performed in the other 262 samples. The clinical notes provided with samples from 138 of the 262 children contained sufficient detail to allow us to evaluate independently the likelihood that the subjects had OI using clinical criteria alone. To be included in this subset of 138 infants, the notes usually had to include a description of the fractures, notation of a family history of OI or frequent fractures, colour of sclerae, presence or absence of Wormian bones, bowing of extremities, osteopenia, and measurement of stature. Some descriptions that did not have all items in place were included if they had a physical examination with a listing of pertinent positives and/or negatives. Additional data collected from the notes provided included involvement of CPS, statements as to probable diagnosis, and the reasons given for testing.

Age distribution in months of children for whom biochemical studies of cultured fibroblasts were completed. All samples studied (open circles, age data available for 238 of the total of 262 children); samples from those with adequate clinical histories for independent analysis (filled squares, age data available for 135 of the 138 children). Seven children with ages greater than 36 months are not included in this figure.
Biochemical studies
Cultured fibroblasts were sent to us from the referring hospital or were grown in our laboratory from skin biopsies, as previously described.22 Collagenous proteins were labelled and analysed by polyacrylamide gel electrophoresis as previously described.22 The resulting autoradiographs were analysed for the amounts and mobilities of the chains of type I procollagen and collagen.
Characteristically, cells from subjects with OI type I make about half the normal amount of type I procollagen, but the chains have normal electrophoretic mobilities.20 In contrast, cells from subjects with OI type IV and many forms of OI type III make normal amounts of type I procollagen but the electrophoretic mobilities of the chains of type I procollagen are abnormal and the efficiency of secretion of the abnormal molecules is reduced.20
Analysis of expression of COL1A1 alleles
In some cases there was a decreased amount of type I procollagen produced, but not enough of a decrease to allow us to confirm a diagnosis of OI. In these instances, DNA was isolated from cultured cells and the regions surrounding a polymorphic MnlI site in the 3` untranslated region (3` UTR) of the COL1A1 gene were amplified and cleaved with MnlI.23 When heterozygous, the two alleles were assayed in cDNA synthesised from mRNA by reverse transcription and amplification of the product by the polymerase chain reaction (RT-PCR) followed by digestion of the product with the same enzyme.24
RESULTS
Biochemical studies of collagen production and structure
In our studies of these 262 infants and children, we identified 11 in whom the diagnosis of OI was clear. Among these, cells from six synthesised about half the normal amount of normal type I procollagen, consistent with the diagnosis of OI type I. Cells from the remaining five infants synthesised some abnormal molecules, consistent with the diagnosis of OI type IV or OI type III. In another 11 we could not exclude OI, usually because we could not be sure if the amount of type I procollagen synthesised was in the normal range. In these instances, when we tried to determine if both COL1A1 alleles were expressed in equal amounts, the polymorphic marker used (an MnlI site in the 3` untranslated region) was homozygous in genomic DNA and thus non-informative.
Clinical correlations
The subgroup of 138 well described infants included nine of the 11 infants in whom we identified OI (six with biochemical findings of OI type I and three with the abnormal findings seen in subjects with OI type III or IV), six of the 11 in whom we could not exclude OI, and 123 in whom studies were normal (fig 3). The referring physicians, 90% of whom had training in clinical genetics, suspected that the children had OI in 10 of these 138 infants. They had designated six of the nine we showed to have OI as affected and thought that four of the 123 for whom normal results were obtained also had OI (table 1). In our review of the data provided for these latter four patients, the diagnosis of OI was suspected on the basis of fractures and “blue tinged” sclerae in three of four. In one instance, the basis of the diagnosis was unclear. Blue sclerae were noted in almost half of the 138 children and are recognised as an age dependent normal finding.13,14
Correlation between referring clinical diagnoses and biochemical diagnoses

{kind=link}
{kind=link}
{kind=link}
Outcome of study of the 138 samples with adequate clinical information of the total of 262 samples submitted over a 38 month period for analysis.
For this group of 138 patients we could find no combination of additional clinical signs (for example, blue sclerae, short stature), associated history (family history or history of injury), or radiological features (nature and location of the fractures or Wormian or bowed bones), which, when combined with fractures, discriminated all affected infants from those we determined to be unaffected (table 2). In part, this may have been because we had incomplete clinical information and did not have x rays to review for most infants.
Clinical features and findings of nine subjects (with adequate clinical documentation) identified with abnormal type I procollagen
One unanticipated feature of the group we studied was the higher proportion (23/138, 16.7%) of infants born at 36 weeks' gestation or less when compared to the US norm (9.0%, χ2=7.67, p=0.01) (US Vital Statistics (http://www.cdc.gov/nchs/data)). Twelve were born before 32 weeks' gestation and five had some degree of developmental impairment. Six were part of multiple births. Cells from one infant born at 34 weeks' gestation synthesised somewhat less type I collagen than control cells and we were unable to exclude the diagnosis of OI type I. Cells from the rest synthesised normal amounts of type I collagen. The overall proportion of twins in this group (11/138, 7.9%) was greater than the expected 2.8% in the US population (χ2=17.68, p=0.001) (US Vital Statistics (http://www.cdc.gov/nchs/data)), and accounted for most of the prematurity in this group so that twinning and prematurity were not independent variables.
In the group of children whose cells made normal collagens, 36 of the 138 (26%) had clear evidence of additional trauma. There were four who had died, one who was found abandoned, 11 who had intracranial haemorrhages, five who had retinal haemorrhages, and 14 who had bruises, burns, bites, lacerations, or clear evidence of sexual abuse. For an additional eight children, there had either been previous abuse of children in the family or fractures stopped once the child was removed from the home. This compares to one bloody nose in the OI positive group and one laceration in the mouth of one of the equivocal subjects.
Pattern of referral for testing
Of the 138 infants for whom we had the most complete data, 103 were known to have been referred to Child Protective Services (CPS) and the data were incomplete for the other 35. The court ordered testing for 21 of the 103; in two of them we could not exclude OI. For an additional 24, the court was involved in facilitating testing, although we were unsure of the exact nature of the court's role; none had OI. Among the remaining 58 who had been referred to CPS, seven had OI and in four we could not exclude the diagnosis. The two other children in whom we made the diagnosis of OI were among the 35 for whom we did not know if CPS was involved.
DISCUSSION
In our retrospective study, we identified 11 of the 262 infants (4.2%) at risk to have NAI as affected with OI and could not exclude the diagnosis in an equal number. Six of the 11 with OI were considered to be affected by the referring physicians, three were considered “possibly” affected, and there was no information for the other two. Among those for whom biochemical studies were equivocal, none was thought by the referring physicians to have OI on clinical grounds.
Given the prevalences of OI and NAI in the USA, if children with OI and those thought to have been intentionally injured were combined into a single group, approximately 2-5% of the total would be expected to have OI, approximately the proportion we identified here and in our previous, smaller study.25 The fact that the proportion of children with OI among the total is similar to that expected if there were no preliminary screening on clinical grounds suggests two things. First, given that genetic testing was requested for infants in whom the diagnosis of OI was suspected on clinical grounds, it appears that physicians are compelled to request laboratory confirmation of the OI diagnosis in instances of possible non-accidental injury. Second, it is likely that in the population of children with unexplained fractures, it is difficult to distinguish those with OI on clinical grounds alone in a subset of children, probably because of the age dependent expression of clinical signs like dentinogenesis imperfecta, hearing loss, bone deformity, and short stature. Thus, despite our previous conclusion that clinical examination is likely to provide a sufficient screening tool,25 it now seems clear that some children with OI are not ascertained easily by examination alone and that laboratory testing can be an important element in their identification.
These tests are efficient at identifying those with OI type I and OI type IV, but less so those with OI type III, either because the mutations do not alter protein or because the mutations are in other genes.20 Because the x ray features of OI type III should be readily recognisable in the 0-3 year age group and because those children are readily identifiable by clinical examination, it is children with OI type I or OI type IV that are most likely to be overlooked by examination in the heat of the moment when NAI is considered as the most likely source of the injury. If we assume that the ability to detect biochemical abnormalities in cells from these children is 90% and we identified 11 affected children, then we would have failed to detect one child (or two, if the number “detected” is considered to be 22 instead of 11). Thus the chance that the 240 infants in this study who had normal studies would have OI should be about 1% or less.
For reasons that are not clear, assessment of unexplained fractures has evolved to revolve around the dichotomy of OI and abuse. There are other predisposing genetic factors for bone fragility and fracture that include hypophosphatasia and related disorders,26 disorders of vitamin D metabolism,27,28 and disorders of copper metabolism such as Menkes syndrome,29,30 to name only three additional genetic disorders. The exclusion of OI as a contributing factor does not exclude other factors, although the a priori likelihood of their involvement is not high, because in most instances the other genetic predispositions to fracture are less common than OI.
The relatively high proportion of premature infants in this study reawakens the discussion of other factors that could predispose to fracture in children. Prematurity contributes to fracture predisposition among infants with birth weights less than 2000 g and gestational ages less than 30 weeks,31,32 but most of these fractures occur in the hospital. Only four infants in our study population were born before 30 weeks' gestation suggesting that the prolonged hospitalisation and nutritional concerns of the very premature were limited in this instance. The increased risk for fracture in the premature does not seem to be sustained into childhood. In studies of children under the age of 5 who presented to an emergency room because of fractures, the incidence of prematurity was not higher than seen in children presenting for other reasons who did not have fractures.33 Premature infants, particularly those with developmental and medical concerns, may be members of a population of “disabled” children who are at greater risk for non-accidental injury.34 The precise role that prematurity plays in unexplained fractures, whether it is a primary biological role of bone disease of prematurity or a secondary social association, is unclear and warrants further investigation.
It is difficult to recommend which children seen in the context of an evaluation of fractures warrant further study to determine if they have OI. Our studies failed to identify clear clinical criteria that separated those with OI from those without, perhaps because we did not examine the children ourselves and did not have complete information. On the other hand, none of the children who had other signs of abuse was identified as having OI and this identifies a group for whom additional study is not warranted. Finally, it is likely that the combination of studies of protein production by cultured fibroblasts and molecular genetic analysis of the type I collagen genes will identify a small number of additional children with OI. We are aware that complementary studies have found abnormalities not found by the other method, although the rate seems quite low.
Although the majority of infants we studied were assessed by child protective services (CPS), we know little about how these biochemical test results are used in the civil or criminal justice system. Testing for OI presents an unusual situation in the legal system in that testing one person for the presence of a genetic condition may have a bearing on the health of a parent and, conversely, on the ability of that parent to continue in the household. Few other tests have such a role and it is important that as genetic testing becomes more common, and the use of test results in the civil and criminal justice system becomes more frequent, we develop methods to ensure that the results are used in both a scientifically valid fashion and with compassion.
Acknowledgments
These studies were supported, in part, by a grant from the National Institutes of Health (AR41223). We are indebted to many physicians for providing the data we summarised and assessed in the course of these studies.
Electronic databases and addresses cited: National Child Abuse and Neglect Data system, 1999, http://www.acf.dhhs.gov/programs/cb/publications. Database of Human Type I and Type III Collagen Mutations, http://www.le.ac.uk/genetics/collagen. US Vital Statistics, http://www.cdc.gov/nchs/data.