Article Text

Abstract
BACKGROUND We have sought to establish the prevalence of goitre within a Pendred syndrome (PS) cohort and to document the course of thyroid disease in this patient group. As part of a genetic study of PS we have assessed 57 subjects by perchlorate discharge test and in 52 (M 21, F 31, age range 9-54 years) a discharge of radioiodide of >10% was observed.
RESULTS Goitre was present in 43 (83%) of the cohort (28 F, 15 M), generally developing after the age of 10 years, 56% remained euthyroid (age range 9-37 years), and 19 patients (44%) had objective evidence of hypothyroidism, all of whom had goitre.
CONCLUSIONS In summary, thyroid dysfunction in PS is variable and inclusion of goitre as a diagnostic requirement will maintain significant underascertainment. The recent identification of the genetic defect underlying PS is likely to provide an important diagnostic aid in the identification of this disorder and this communication should assist clinicians in identifying deaf patients who ought to be considered for this investigation.
- Pendred syndrome
- thyroid disease
Statistics from Altmetric.com
Inherited deafness is common (approximately 1 in 2000 live births) and approaches to the investigation of hearing impaired children are varied.1 The inherited hearing disorders are generally classified as syndromic, in which case there will be other clinical signs accompanying the deafness and signalling a possible unifying diagnosis and inheritance pattern, or non-syndromic, in which deafness is the only abnormality. Most clinicians hope to recognise the more prevalent syndromic forms of deafness from the constellation of clinical findings. Recognition of a syndrome will guide future investigation of that patient, avoid more general approaches to investigation which might be appropriate when a specific diagnosis is not identified, and provide a basis for genetic counselling in the family. However, syndrome recognition relies upon the consistency of clinical findings in a particular diagnosis. This communication details our experience of the clinical findings of thyroid disease in Pendred syndrome and signals some potential pitfalls in making this diagnosis.
The definition of Pendred syndrome (PS) has continued to be determined by the association of goitre (resulting from thyroid cell hyperplasia) with sensorineural deafness. The association of these two clinical features was observed by Vaughan Pendred2 in 1896, who described a family comprising 10 sibs in which two sisters had deafness and large goitre in an area where goitre was not endemic. In 1927, Brain3 reported several further cases and postulated that not only was the expression of goitre and deafness the result of the same abnormal gene in homozygous form, but also that the goitre may be associated with a specific inborn error of thyroxine synthesis. Fraser4 defined the place of Pendred syndrome as “a numerically important component of the heterogeneous entity of congenital deafness... characterised by an invariable association with an inborn error of thyroxine synthesis at the level of organic incorporation of trapped iodide in the thyroid gland.” Based on his survey of more than 3500 patients with profound childhood deafness, he estimated that PS might comprise 4.3-7.5% of all causes of childhood deafness.
The deafness is sensorineural and occasionally associated with disturbed vestibular function, while characteristic developmental anomalies of the cochlea in the form of dilated vestibular aqueducts or Mondini defect are recognised as associated features and can be shown radiologically.5 6 However, while the hearing loss is a constant feature of PS, the goitre has been reported to vary significantly in size within families and may even be absent.4 7-9 Despite the recent identification of the gene for PS, the molecular basis of the association between hearing loss and a defect in the organification of iodide remains unclear.10
The disorder is characterised by the incomplete discharge of radioiodide from a primed thyroid following perchlorate challenge.11 In a normally functioning thyroid gland inorganic iodide, having been trapped, is immediately organified by binding to thyroglobulin. Perchlorate unmasks defects of organification by provoking the discharge of inorganic iodide from the gland, and in patients with PS between 15 and 80% of accumulated iodide is discharged on administration of perchlorate, compared to less than 10% in normal controls. However, both false positives and false negatives have been reported.9 12 13
Since goitre often prompts the diagnosis, leading to appropriate investigation of patients and subsequent confirmation of the condition, prevailing published reports are not well informed as to the prevalence of thyroid disease in Pendred syndrome. Moreover, variability in the clinical features of the thyroid element of the syndrome is well established and it is possible that the condition is underdiagnosed.9 We have, in the course of a genetic study of PS, had the opportunity to assemble data on a large series of cases, most of whom have been systematically evaluated, clinically, biochemically, radiologically, and by perchlorate challenge. With the exception of Fraser’s study of 334 cases in 1965,4 there are few published data relating to the presentation and natural history of thyroid disease in PS which would assist clinicians in identifying patients with this condition. The present study aims to further our understanding of these issues.
Methods and results
Forty nine families comprising 72 deaf subjects were ascertained from all over Great Britain, among whom there were 20 sib pairs. Criteria for inclusion in the affected cohort were (1) sensorineural deafness, (2) a positive result on perchlorate discharge testing of the thyroid gland (>10% discharge), and (3) the absence of antithyroid antibodies. Patients were derived from three sources. (1) Patients noted to have a hearing loss associated with a goitre or thyroid dysfunction were referred by medical colleagues through clinics in audiological medicine, general paediatrics, endocrinology, and general practice. (2) Patients were identified through Departments of Radiology whose deafness was associated with characteristic radiological malformations of the inner ear known to be associated with PS. (3) Deaf sibs or probands of affected subjects were also ascertained through referrals from medical colleagues, enquiries into the family history, or information volunteered by affected subjects or their families.
Fifty-seven subjects were investigated with a perchlorate test and our observations are confined to this group. Of these, 52 (91%) had results consistent with a discharge of radioiodine of >10% following administration of the perchlorate ion. The majority of tests (46 of 57) were carried out at St Bartholomew’s Hospital according to a standard protocol, while data from tests performed at other centres in accordance with a different protocol were reviewed. Five patients were found to have negative perchlorate discharge tests despite other findings, radiological or familial, suggesting PS, as shown in table1.
Characteristics of patients with negative perchlorate tests
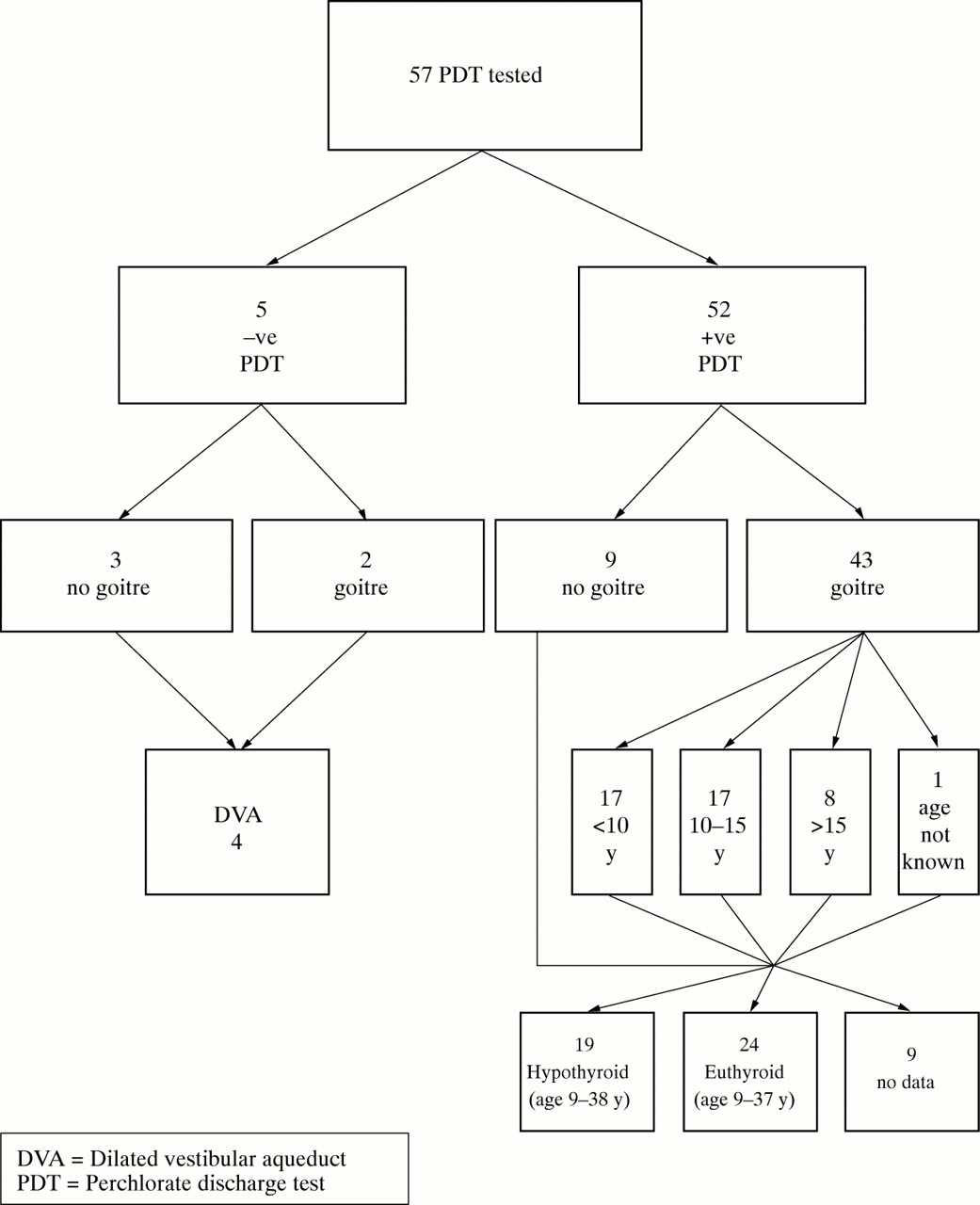
The 52 subjects who satisfied the inclusion criteria were reviewed with specific reference to thyroid disease. Affected subjects ranged in age from 9 to 54 years (31F, 21M) and 21 cases (40%) arose sporadically, while 31 (60%) were familial. The presence of goitre in an affected subject was ascertained by studying documentation in their case records of a thyroid examination performed by a paediatrician or endocrinologist, in which the presence or absence of a goitre was noted. The age at which the goitre first became apparent to parent, patient, or clinician was also ascertained by studying case records, and discussion with the patient or parent. The age of onset of goitre remained unknown in one patient whose case notes were incomplete and attempts at further ascertainment were not possible. The breakdown of data are outlined in fig 1.
{kind=link}
Breakdown of data.
Goitre was present in 43 of 52 (83%) cases, 15 male and 28 female. In 17 (33%), the goitre was noted before the age of 10 years, in 17 (33%) between the ages of 10 and 15 years, and after the age of 15 years in eight cases (16%).
In an effort to ascertain thyroid status before any therapeutic intervention, biochemical records and correspondence were reviewed and contact made with relevant clinicians. No data were identifiable for nine patients (17% of the cohort); 19 of the remaining 43 subjects (44%) of age range 9-38 years were identified as hypothyroid, and 24 of 43 (56%) of age range 9-37 years identified as euthyroid on the basis of biochemical data or a clear statement of thyroid status by a clinician in the correspondence. The youngest age at which hypothyroidism was diagnosed was 15 days following neonatal screening.
Case records were reviewed and enquiries made regarding the type of intervention received by affected patients. Sixteen cases received no intervention, 36 patients received medical treatment in the form of thyroxine, and of these 15 of 43 with goitre (35%) progressed to surgical intervention. Nineteen patients were hypothyroid (44%), all of whom had goitre. Clinical practice varied widely, some patients being treated surgically before the instigation of thyroxine medication, others having a trial of medication before surgery.
Discussion
Goitre was an important element in the first clinical description of this condition by Pendred2 in 1896 and its presence remains a cornerstone of the diagnosis. Fraser4 described how the clinical thyroid status in PS “is subject to as great or even greater variation as the auditory loss” and that “all sorts of variations from gross goitrous cretinism to a barely detectable enlargement of the thyroid gland may occur”. Patient ascertainment in that survey was based, for the most part, on clinical examination and this report constitutes the first in which clinical features of thyroid disease in PS have been evaluated in the context of all patients having had perchlorate testing as part of the protocol.
The sex ratio in favour of females in the present series and the greater proportion of familial cases is consistent with previous documentation of this autosomal recessive condition. While Johnsenet al 14 suggested that up to 50% are hypothyroid, Fraser,4 in a far larger study, observed most cases to be euthyroid, and the present study supports the variability of thyroid status with definite biochemical evidence of hypothyroidism in 19 cases (44%). In our experience hypothyroidism has only been observed in the presence of goitre. The youngest patient was diagnosed following neonatal screening although she became biochemically euthyroid over time, and her case was initially misdiagnosed as transient neonatal hypothyroidism before she subsequently relapsed. This phenomenon has been well described by Coakley et al,7 who studied the incidence of PS following neonatal thyroid screening and showed an incidence of 1 in 153 000 births, pointing out that as the thyroid enlargement in this condition is also subtle, these children may easily be lost to follow up. They emphasise the need for repeat thyroid function studies at intervals in any so called case of transient hypothyroidism of uncertain aetiology and that the first audiological assessment should be undertaken in the first 4-6 weeks of age in patients in whom there are strong reasons for suspecting PS.
Fraser4 described females as being more frequently and severely affected with goitrous enlargement and this again is supported by a sex incidence of goitre of M15:F28, and there being a greater proportion of females comprising those cases that came to surgery (11F:4M), while those requiring repeat surgery were all female (n=5). Apart from a possible increased cosmetic awareness among females, the reasons for this remain unclear.
In this series, 21 of 52 patients included (40%) were controlled by medical treatment with thyroxine only. However, 15 patients (28% of the total or 35% of the 43 with goitre) progressed to surgery, which is perhaps a higher proportion than expected in view of Fraser’s observation that response to treatment by exogenous hormones is most satisfactory. However, no data were available regarding the age at which treatment was initiated, and it is possible that regression of the goitre may occur if treatment is started early in life.4 Twenty two operations were carried out, seven of which were repeat procedures, supporting Fraser’s observation that there is a poor response to thyroidectomy owing to regrowth of the remnant.
Our data emphasise that the goitre in PS appears most commonly in the second decade of life with almost 70% in our series having no clinical evidence of goitre at the age of 10 years. In very few cases the goitre is congenital or appears in early infancy. The patient age at onset of the goitre was documented on the basis of when the swelling was first apparent to the patient, parent, or clinician and it is recognised that this observation may be subject to interobserver variability in the same case and between cases.
As described previously, the diagnosis of PS is an important one to make, particularly at an early stage, in order to facilitate patient management and appropriate genetic counselling of the family. The genetic locus responsible for PS has recently been localised to 7q31 and mutations identified in an ion transport gene,10making molecular diagnosis possible. However, while it is hoped that mutation detection will allow confirmation of the diagnosis in affected patients, its success as a diagnostic tool, as with any form of genetic testing, is highly dependent on clinical suspicion based on an accurate knowledge of clinical criteria and their spectrum of variability. As the data we present emphasise, other avenues of investigation for Pendred syndrome, radiological, biochemical, and molecular, are appropriate in the consideration of the non-goitrous deaf child if ascertainment of PS is to be improved.
Acknowledgments
Drs Reardon and Trembath are pleased to acknowledge grant assistance from Action Research and from Defeating Deafness, the Hearing Research Trust.