Article Text

Abstract
Inflammatory bowel disease (IBD), encompassing Crohn's disease and ulcerative colitis, has multifactorial aetiology with complex interactions between genetic and environmental factors. Over 150 genetic loci are associated with IBD. The genetic contribution of the majority of those loci towards explained heritability is low. Recent studies have reported an increasing spectrum of human monogenic diseases that can present with IBD-like intestinal inflammation. A substantial proportion of patients with those genetic defects present with very early onset of intestinal inflammation. The 40 monogenic defects with IBD-like pathology selected in this review can be grouped into defects in intestinal epithelial barrier and stress response, immunodeficiencies affecting granulocyte and phagocyte activity, hyper- and autoinflammatory disorders as well as defects with disturbed T and B lymphocyte selection and activation. In addition, there are defects in immune regulation affecting regulatory T cell activity and interleukin (IL)-10 signalling. Related to the variable penetrance of the IBD-like phenotype, there is a likely role for modifier genes and gene–environment interactions. Treatment options in this heterogeneous group of disorders range from anti-inflammatory and immunosuppressive therapy to blockade of tumour necrosis factor α and IL-1β, surgery, haematopoietic stem cell transplantation or gene therapy. Understanding of prototypic monogenic ‘orphan’ diseases cannot only provide treatment options for the affected patients but also inform on immunological mechanisms and complement the functional understanding of the pathogenesis of IBD.
- INFLAMMATORY BOWEL DISORDERS
- IBD - GENETICS
- IMMUNODEFICIENCY
- INFANT GUT
Statistics from Altmetric.com
Monogenic disorders and inflammatory bowel disease
The aetiology of inflammatory bowel disease (IBD), encompassing the main disease subgroups Crohn's disease (CD) and ulcerative colitis (UC), is complex and multifactorial.1 ,2 Recent genetic association studies within large IBD cohorts have identified 163 genetic loci.3 These 163 loci in total explain 13.6% of CD and 7.5% of UC total disease variance3 and the majority of those loci contribute only little towards the explained heritability. Genes within IBD associated genetic loci highlight the importance of epithelial barrier defects, innate recognition and response towards the microbiota, in particular the role of autophagy and T helper cell (Th) 17 type driven dysregulated adaptive immune responses. Whereas the functional role of some of the most strongly associated genes such as NOD2, ATG16L1 or IL23R becomes increasingly clear, the functional impact of most key candidate genes within IBD loci is currently not understood.1 ,2
In addition to IBD there are an increasing number of monogenic diseases that can present with IBD-like immunopathology. The prevalence of these monogenic diseases is very low compared with common intestinal infections, coeliac disease or IBD (figure 1). As a group, these monogenic defects are associated with high morbidity and even mortality. Due to their clear functional impact, these prototypic ‘orphan’ diseases can inform on functional mechanisms that predispose to intestinal inflammation. Indeed, there is increasing awareness that some aspects of the immune dysfunction in IBD may have similarities to those seen in rare monogenic diseases.4–7

Estimated prevalence of monogenetic disorders that can present with inflammatory bowel disease (IBD)-like immunopathology in comparison with IBD, coeliac disease and common enteric pathogens. Prevalence of monogenic disorders compared with IBD89 and coeliac disease97 in Western countries. CD, Crohn's disease; CVID, common variable immunodeficiency; IL, interleukin; IPEX, immune dysregulation, polyendocrinopathy, enteropathy, X linked; UC, ulcerative colitis.
In the following, I focus on monogenic disorders based on a selective literature search, including Pubmed and Online Mendelian Inheritance in Man (OMIM) database search. For the sake of clarity, intestinal inflammation in Mendelian disorders is regarded as IBD-like (CD-like, UC-like) immunopathology even if the reported endoscopic and histological picture does not allow a distinction to IBD. I compare the immunological mechanisms of prototypic orphan diseases and discuss implications for the understanding of IBD. Animal data are only provided in selected cases when regarded helpful for the understanding of possible functional mechanisms.
Monogenic defects are more frequent in patients with very early onset of IBD-like inflammation
CD and UC have a peak onset between 20 and 40 years of age (figure 2). Only a minor fraction of all IBD patients develop intestinal inflammation within the first 6 years of life, and onset in the first year of life is extremely uncommon.8–12 A distinct phenotype in children with very early onset IBD is indicated by a high proportion of pancolitis and severely ulcerating and fistulising disease develops in subgroups of patients.13 ,14 A high percentage of patients in this age group cannot be classified as CD or UC. Indeed, the frequency of unclassified IBD (IBDu or IBD ‘yet to be classified’ or indeterminate colitis) is less than 5% in the adult IBD population but rises to 21% in the group under 7 year of age and up to 34% in those aged under 3 years of age.15
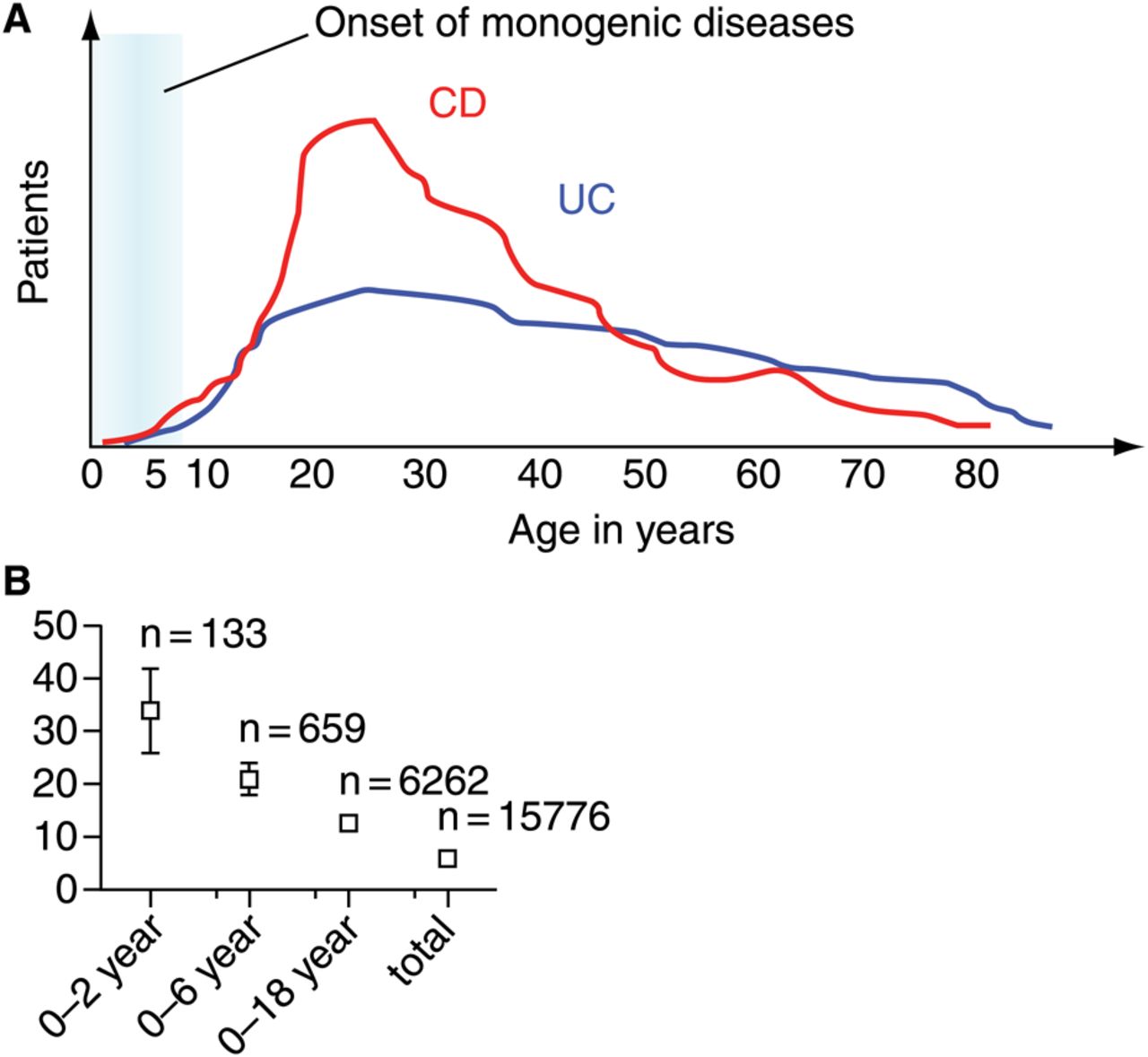
Age distribution at diagnosis for Crohn's disease (CD) and ulcerative colitis (UC) and frequency of unclassified/indeterminate colitis within different age groups. (A) Age of diagnosis of CD and UC. The schematic represents a representative population based inflammatory bowel disease (IBD) cohort of 1527 patients (modified after Petritsch and colleagues98). The blue field highlights the time interval for the onset and diagnosis of IBD-like symptoms in a substantial proportion of monogenetic disorders (for further information see table 1). (B) High frequency of indeterminate/unclassified colitis in very young children. The percentage of children with indeterminate/unclassified colitis is based on a meta-analysis of published studies (modified after Prenzel and Uhlig15). Numbers of patients per group are indicative for the restricted information basis in very early onset IBD. 95% confidence interval are provided.
It has been suggested that children with early and very early onset of IBD-like inflammation may not only represent a distinct phenotype but also a distinct genetic architecture.16–18 Children with very early onset of intestinal inflammation that are currently classified as ‘classical’ IBD may harbour either specific polymorphisms associated with early onset or might have a particular high load of genetic variants that confer susceptibility to IBD. A proportion of those patients with early and particularly very early onset IBD (onset <6 year of age) suffer from monogenic disorders (see table 1).
Genetic defects associated with IBD-like immunopathology
Monogenic defects represent key functional nodes within inflammatory networks
Only three genes are shared among the 40 monogenetic defects with IBD-like phenotype selected in this review with the IBD key genes clustering within 163 genetic loci (figure 3A). This is a surprisingly low direct overlap. However, proteins encoded by a large proportion of monogenic disorders have direct or indirect interaction partners with genetic variants that confer genetic susceptibility to IBD. Indeed, there are several protein–protein interaction networks that indicate common signalling cascades that confer susceptibility to intestinal inflammation (figure 3B, C).
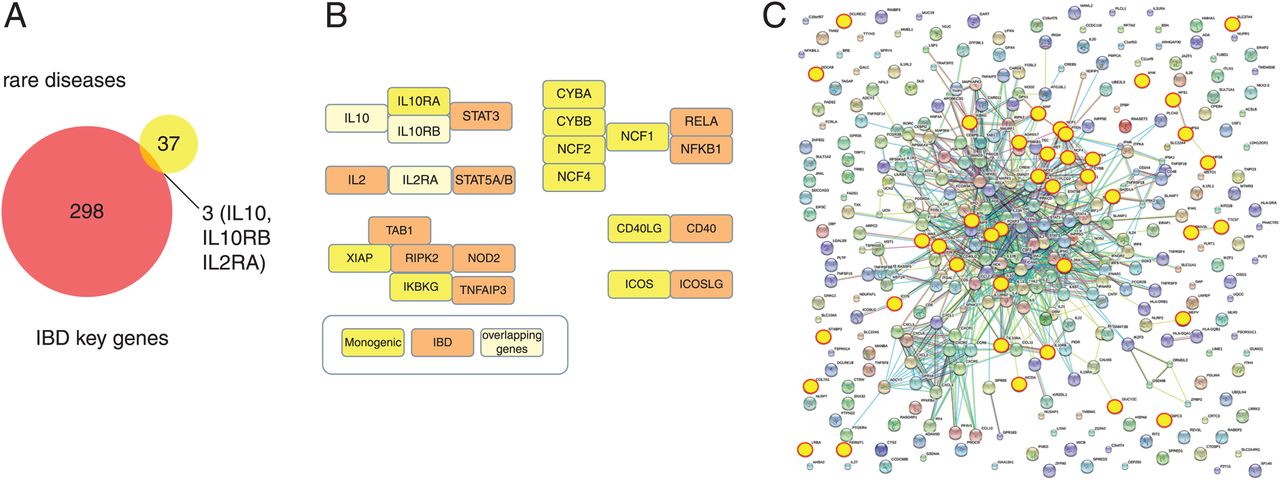
Pathway interaction between key genes associated with inflammatory bowel disease (IBD) and IBD-like monogenic disorders. (A) Comparison between key genes within 163 IBD loci3 and 40 genes associated with IBD-like monogenic disorders. The proportional area Venn diagram was constructed using Biovenn (http://www.cmbi.ru.nl/cdd/biovenn/). (B) Examples for signalling pathways and protein network interactions that are involved in the pathogenesis of intestinal inflammation. Shared genes (IBD associated and IBD-like monogenic genes) as well as IBD associated only and IBD-like monogenic only genes are shown separately. (C) Functional gene network analysis. All 301 key IBD genes and IBD-like monogenic genes were analysed by interaction network using String database (http://string-db.org/). Monogenic genes, highlighted in yellow, are largely positioned within the IBD interaction network and the only exceptions are not connected according to the current information base.
There are several groups of monogenic diseases, including chronic granulomatous disease (CGD), interleukin (IL)-10 signalling defects, Hermansky–Pudlak syndrome, Wiskott–Aldrich syndrome, immune dysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome or mevalonate kinase deficiency, where IBD-like pathology has been very convincingly shown in a large number of genetically defined patients. However, case series or even single case reports can start to inform on mechanisms when genetic findings are linked to strong phenotype segregation within families or when genetics is supported by functional human data or animal models of IBD.
According to plausible functional mechanisms, the 40 selected monogenetic defects can be grouped within subtypes:
-
epithelial barrier and epithelial response defects
-
neutropenia and defects in phagocyte bacterial killing
-
hyper- and autoinflammatory disorders
-
disorders affecting T and B lymphocyte selection and activation
-
immune dysregulation disorders with defects in negative control of innate and adaptive immune responses
These mechanisms partially overlap and are summarised in figure 4 and table 1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Model of key intestinal defence functions and cellular immune regulation components that are impaired in monogenic disorders with inflammatory bowel disease (IBD)-like phenotype. Genetic defects that affect epithelial barrier and response, neutrophil numbers, phagocytosis and bacterial killing, as well as gut innervation lead to increased intestinal bacterial translocation across the epithelial barrier and bacterial survival. Translocated bacteria can trigger a sustained innate hyper- and autoinflammatory immune response which cannot be controlled. A selection and activation defect affecting T and B cell responses may contribute to colitis via autoimmunity and/or modified adaptive immune response towards the bacterial flora. Immune polarisation towards Th1, Th2 or Th17 responses depends on the underlying genetic defect and the sustained innate response towards enteric microbiota. Lack of functional FOXP3+ regulatory T cells or defects in anti-inflammatory interleukin (IL)-10 signalling can result in intestinal inflammation due to lack of regulation of the proinflammatory response. There are several feedback and amplification mechanisms. Autoimmunity or haemophagocytic events may participate by inducing neutropenia, lymphopenia or epithelial barrier destruction. A further amplification mechanism is potentially the change in the bacterial microbiota composition secondary to the inflammatory process or changes in intestinal innervation and transport function.
Epithelial barrier and epithelial response defects
In the intestine, a single epithelial cell layer separates potentially pathogenic bacteria in the luminal content from immune cells in the lamina propria. It is therefore not surprising that defects affecting epithelial barrier function and epithelial response to pathogens can predispose to intestinal inflammation. Among the prototypic disorders that primarily affect epithelial barrier function and can present as IBD-like immunopathology are dystrophic epidermolysis bullosa and Kindler syndrome19 but also familial diarrhoea due to activating dominant guanylate cyclase C mutation.20 X linked ectodermal dysplasia and immunodeficiency, and ADAM-17 deficiency are complex diseases but there is evidence that the IBD-like phenotype is largely due to epithelial dysfunction. In addition, there is a group of Hirschsprung's disease patients where enterocolitis is caused by increased bacterial translocation as a consequence of dysfunctional intestinal innervation due to a germline RET mutation.21 ,22
X linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by hypomorphic mutations in IKBKG encoding nuclear factor kB essential modulator protein (NEMO). Enterocolitis with villous atrophy and epithelial cell shedding are typical findings.23 Allogeneic bone marrow transplantation corrected the immunodeficiency but failed to correct the colitis.24 Similarly, mouse data suggest that epithelial specific NEMO deficiency leads to intestinal inflammation,25 suggesting that NEMO deficient epithelial rather than haematopoietic cells cause IBD-like inflammation. These studies suggest that nuclear factor κB signalling in epithelial cells is required to prevent excessive epithelial cell apoptosis and to maintain epithelial barrier integrity.
ADAM-17 encodes a disintegrin and metalloprotease that cleaves diverse substrates, including tumour necrosis factor α (TNFα), L-selectin or epidermal growth factor (EGF) receptor ligands from the cell membrane.26 Patients with defects in ADAM-17 presented with neonatal onset of non-bloody later bloody diarrhoea. ADAM-17 deficient peripheral blood mononuclear cells secreted reduced level of TNFα but not IL-1β, IL-6 or interferon γ on stimulation compared with controls.27 Although this might explain an element of immunodeficiency, it is likely that the epithelial dysfunction mediates intestinal inflammation in those patients. Indeed, mice with reduced ADAM17 activity have increased intestinal permeability and increased susceptibility to dextran sulfate sodium colitis.26 Treatment of ADAM17ex/ex mice with the EGF receptor ligand, transforming growth factor α, reduced dextran sulfate sodium induced pathology.26 These results indicate that among the many functions of ADAM-17 in inflammation and tissue remodelling, susceptibility to colitis is an epithelial intrinsic defect due to the inability to activate EGF receptor mediated epithelial stress response and integrity.
The importance of the epithelial barrier function for the pathogenesis of IBD was similarly illustrated by genetic association studies highlighting the role of genes including HNF4A, GNA12 or XBP1.3 ,28 ,29 It should be noted that similarities in the pathomechanisms between monogenic rare diseases and IBD are not only revealed by genetic variants that affect epithelial barrier function. Indeed, IBD-like immunopathology was not only described in patients with dystrophic epidermolysis bullosa due to defects in collagen VII (COL7A1)19 but also in a group of IBD patients with autoimmunity towards this collagen. Those patients with epidermolysis bullosa aquisita can present with a CD dominant phenotype (reviewed in Reddy and colleagues30), highlighting the functional importance of collagen VII for maintaining intact epithelial barrier function in two independent settings.
Bacterial handling by neutrophil granulocytes
Neutrophils are part of the intestinal innate barrier.31 Several classical monogenic disorders, including CGD, glycogen storage disease type 1b, congenital neutropenia or leucocyte adhesion deficiency 1, present with neutropenia and functional neutrophil defects. Several other genetic defects, including WAS, LRBA, BTK, CD40LG or FOXP3 defects, display a neutropenic component, which is partially autoimmune. On the other hand, not all forms of neutropenia are associated with IBD-like pathology. This suggests that even low numbers of functional granulocytes can largely protect from intestinal inflammation.
CGD is a prototypic example of the important defence function of intestinal phagocytes in particular granulocytes. The phagocyte NADPH oxidase (phox) complex generates the microbicidal ‘respiratory burst’ that is required to kill ingested intracellular microbes. The phox complex consists of the cell membrane integrated gp91-phox (CYBB) and p22-phox (CYBA) subunits, the cytosolic subunits p47-phox (NCF1) and p67-phox (NCF2) as well as p40-phox (NCF4). Mutations in any of the five phox complex molecules leads to immunodeficiency and can cause intestinal inflammation.32–34 Interestingly, a missense variant in NCF2 that affects binding to RAC2 is associated with very early onset IBD.35
CD-like immunopathology has been described in patients with other defects in neutrophil numbers and function such as gluco-6-phosphate-translocase (SLC37A4),36 ,37 glucose-6-phosphatase 3 (G6PC3)38 as well as leucocyte adhesion deficiency 1 (ITGB2).39 ,40 Neutropenia and neutrophil dysfunction in both glycogen storage disease 1b (SLC37A4) as well as glucose-6-phosphatase 3 deficiency (G6PC3) are partially explained by shared metabolic pathway defects that lead to increased neutrophil apoptosis in particular under cellular stress.
Treatment options for neutrophil/phagocyte disorders associated IBD-like pathology involve conventional anti-inflammatory treatment but also anti-TNFα treatment.38 ,41–43 However, anti-TNFα treatment in CGD was associated with increased lethality, suggesting that removal of an additional layer of immunoprotection due to TNFα blockade may give rise to (lethal) infections.42 Granulocyte colony stimulating factor may resolve the neutropenic component in glycogen storage disease type Ib,43 and allogeneic haematopoietic stem cell transplantation resolves the functional defect in CGD (boxes 1 and 2).44
Monogenic disorders associated with IBD-like intestinal inflammation
-
A spectrum of rare (‘orphan’) monogenic disorders confer increased susceptibility to IBD-like intestinal inflammation.
-
There are few common genes between the group of IBD associated genes that cluster within 163 IBD loci and monogenic disorders with IBD-like immunopathology but there are several shared immunological pathways.
-
Most monogenic disorders have an onset of colitis symptoms during early childhood.
-
Variable penetrance of the IBD phenotype suggests a role for modifier genes and/or gene–environmental interactions.
Treatment options
-
Conventional therapy used for the treatment of IBD including exclusive nutritional therapy, anti-inflammatory and immunosuppressive treatment is only partially effective.
-
Anti-TNFα treatment can be helpful in a variety of monogenic diseases. In CGD, anti-TNFα treatment has been effective for treating IBD-like pathology but might be associated with increased mortality.
-
Anti-IL-1β treatment can be effective for diseases with strong autoinflammatory components.
-
Bone marrow/haematopoietic stem cell transplantation is a treatment option for multiple diseases, including IPEX, CGD, XIAP and IL-10 signalling defects. In case of NEMO deficiency, bone marrow transplantation can reconstitute the systemic immunodeficiency but may not cure colitis.
-
Gene therapy was applied to treat WAS associated immunodeficiency and colitis.
Interestingly, patients with CD may have specific defects in proinflammatory macrophage cytokine secretion, leading to impaired neutrophil accumulation.45 Subcutaneous injection of Escherichia coli leads to reduced neutrophil accumulation and reduced bacterial clearance in CD patients but not in UC patients or in healthy controls, suggesting that CD patients may have a specific bacterial clearance defect.45 In contrast, patients with UC present with increased numbers of apoptosis resistant activated intestinal neutrophils, which invade the lamina propria as well as the epithelium (cryptitis) and form crypt abscesses.31 ,46 In summary, this suggests that functional defects in neutrophil accumulation or function may increase susceptibility to CD-like intestinal inflammation whereas increased numbers of activated neutrophils may contribute to established inflammation, in particular in UC lesions. Consequently, boost of granulocyte activity in CD patients during remission might be a therapeutic strategy. Whether short term targeting of granulopoiesis or blockade of neutrophil accumulation in the gut could be beneficial during ongoing inflammation needs to be shown.
Hyper- and autoinflammatory disorders
Several syndromes with hyper- and autoinflammatory defects predispose to IBD-like intestinal inflammation. This heterogeneous group of diseases includes autoinflammatory disorders such as mevalonate kinase deficiency, phospholipase Cγ2 defects or familial Mediterranean fever, as well as Hermansky–Pudlak syndrome (types 1, 4 and 6), X linked lymphoproliferative syndrome types 1 and 2 or familial haemophagocytic lymphohistiocytosis type 5 (see table 1).
Among the autoinflammatory disorders, mevalonate kinase deficiency is most strongly linked to IBD-like immunopathology.47 Either increased mevalonate level or decrease of non-sterol isoprenoid end products result in increased activation of caspase 1 and subsequent IL-1β activation.48 Consequently, antagonists to IL-1 can induce complete or partial remission in the majority of patients.47 ,49
X linked lymphoproliferative syndrome 2 is caused by defects in the XIAP gene. Very early onset IBD with severe fistulising perianal colonic phenotype affects about 20% of patients with XIAP defects.50–53 XIAP is involved in NOD2 mediated nuclear factor κB signalling, and several CD susceptibility genes are binding partners for XIAP (figure 3B), suggesting that XIAP is a key node in this inflammatory network.
A complication of hyperinflammatory defects is associated with macrophage and adaptive immune activation. Haemophagocytic lymphohistiocytosis is a potentially lethal hyperinflammatory process caused by several genetic defects, including STXBP2, SH2D1A and XIAP defects.54 Associated with a serum cytokine storm, macrophages become inappropriately activated and phagocytose bone marrow derived cells. Interestingly, haemophagocytic lymphohistiocytosis can be similarly found in a subgroup of therapeutically immunosuppressed paediatric and adult IBD patients.55 ,56 In both the hyperinflammatory monogenic disorders as well as in immunosuppressed IBD patients, the haemophagocytic episodes are often triggered by viral infections, such as Epstein–Barr virus or cytomegaly virus.
Complex defects in T and B cell selection and activation
IBD-like immunopathology has been described in patients with a number of monogenic B cell defects, Wiskott–Aldrich syndrome,57 Omenn syndrome58 or PTEN hamartoma tumour syndrome,59 that have complex alterations in T and/or B cell selection and activation.
Defects that affect B cell numbers and antibody production
Several monogenetic diseases with reduced B cell numbers and antibody production are associated with IBD-like immunopathology. This group includes subsets of common variable immunodeficiency (CVID), hyper-IgM syndrome and agammaglobulinaemia (see table 1).
Despite the classical recognition as B cell defects, there are good reasons to believe that neither the pure absence of B cells nor defects in antibody production itself are linked to the colitogenic effects in those disorders.
The association between common variable immunodeficiency and IBD-like immunopathology is well recognised.4 ,60 In a large cohort of 473 patients, 20 developed IBD (CD, UC, ulcerative proctitis) and a further six presented with no further characterised idiopathic mucosal inflammation.60 However, CVID has a complex and heterogeneous genetic basis, and the known monogenic CVID defects can only explain a minority of patients. Furthermore, most reports on CVID associated IBD-like pathology do not describe specific mutations. Interestingly, the two CVID defects that are linked with IBD-like pathology have non- B cell specific defects. Deficiency in the inducible T cell co-stimulator (ICOS), which is primarily expressed on T cells, is the underlying cause of CVID type 1.61 Analysis of the T cell compartment of one patient with IBD-like pathology and autoimmunity showed a decreased percentage of CD4 central and effector memory T cells, impaired cytokine polarisation and a reduced proportion of regulatory T cells.62
Lipopolysaccharide responsive beige-like anchor protein (LRBA) deficiency, the cause of CVID type 8, can cause large bowel lymphocytic colitis or CD-like pathology.63–65 LRBA is expressed not only by B cells but also by several other cell types and tissues.65 CD19 B cells from peripheral blood of LRBA deficient patients show increased apoptosis and a defect in autophagy.65 This is interesting as genetic association studies have linked IBD, in particular CD, with multiple genes that are involved in the autophagy pathway (in particular ATG16L1 and IRGM1) and it needs to be shown whether the autophagy defect in LRBA deficiency extends to other cell types. In contrast with ICOS and LRBA, CVID subtypes 2 to 7, with defects in TNFRSF13B (TACI), CD19, TNFRSF13C (BAFFR), MS4A1 (CD20), CD81 or CR2 (CD21), are not linked to IBD-like pathology61 (personal communication K Warnatz and B Grimbacher).
Similar to CVID, several subtypes of hyper-IgM syndrome (including defects in CD40LG, AID or IKBKG) can present with IBD-like immunopathology but again the colitogenic defect might be non-B cell intrinsic as the CD40 ligand encoded by the CD40LG gene is largely expressed by activated T lymphocytes; the activation induced cytidine deaminase (AID) gene is not only expressed on B cells but might play a role for IL-10 secreting T cells66 and NEMO deficiency induced IBD-like pathology is most likely due to an epithelial defect (as discussed above).
In addition, it becomes clear that immunoglobulin deficiency alone is not responsible for the increased susceptibility to non-infectious IBD-like intestinal inflammation in patients with B cell defects. Despite its severe lack of immunoglobulins, BTK associated agammaglobulinaemia is less frequently associated with colitis compared with CVID subsets with less severe immunoglobulin deficiency. Furthermore, intravenous immunoglobulin treatment is highly effective in treating infections in children with B cell defects but failed to improve intestinal inflammation in several defects, including BTK, AID and LRBA.65 ,67 ,68
The colitogenic effect in those disorders is therefore either not B cell intrinsic or linked to cellular functions of B cells. It needs to be shown whether—at least in some human B cell defects—intestinal inflammation arises either by the absence of IL-10 producing B cells (B10 cells) or the absence of B cell derived glucocorticoid induced TNFR ligand (GITR ligand) signals. This can affect regulatory T cell subsets, as indicated by animal models.69 ,70
Wiskott–Aldrich syndrome
Defects in the WAS gene lead to a complex immunodeficiency syndrome. UC-like non-infectious colitis manifests in early infancy and can be treated by bone marrow transplantation.57 As a proof of principle, gene therapy using autologous genetically modified haematopoietic stem cells corrected WAS defects and improved symptoms, including haemorrhagic enterocolitis.71 T cell activation, antibody production as well as macrophage or granulocyte activation are affected due to the broad gene expression of the signal adaptor molecule encoded by WAS.57 Mouse data suggest that WAS is particularly essential for the function of CD4+CD25+Foxp3+regulatory T cells.72 Given the broad expression profile of WAS protein within the adaptive immune system, it is surprising that WAS deficiency in innate immune cells plays a dominant role for mucosal immune dysregulation and colitis in a mouse model.73
Omenn syndrome
Omenn syndrome is an autosomal recessive type of severe combined immunodeficiency (SCID) due to DCLRE1C mutations encoding the artemis protein. Defects in V(D)J recombination during T and B cell development cause increased oligoclonal T cells and often largely reduced B cells. A hypomorphic mutation in DCLRE1C allowed residual B and T cell development in a patient with IBD-like immunopathology.58 Further patients with SCID causing genetic defects might develop severe intestinal inflammation due to colitogenic T cells with a restricted repertoire or—at least in some patients—due to maternal lymphocytes causing a intestinal graft versus host disease.
Regulatory T cells and immune regulation
X linked immune dysregulation, polyendocrinopathy, enteropathy
Immune dysregulation, polyendocrinopathy, enteropathy, X linked syndrome (IPEX) represents a prototypic defect in the natural regulatory T cell compartment, leading to immunodeficiency and autoimmunity, but also enteropathy and colitis.74 ,75 A substantial proportion of IPEX patients have defects in the transcription factor FOXP3 that determines the fate and immunosuppressive activity of natural and a part of induced regulatory T cells.74 ,75 The important role of IL-2 signalling in regulatory T cells was indicated by IPEX-like immune dysregulation in a patient with IL-2 receptor α chain defect (IL2RA encoding CD25).76
In patients with IBD, regulatory T cells are present and accumulate in the inflamed intestine.77 ,78 Key autoimmunity features typical for IPEX and IPEX-like patients are absent in IBD patients, indicating that functional regulatory T cells accumulate but cannot control immunopathology in IBD patients.
IL-10 signalling defects
Although the essential function of IL-10 signalling in intestinal immune homeostasis has been recognised in animal models for 20 years,79 the impact of this pathway for human intestinal inflammation has only recently been fully appreciated due to the discovery of monogenic defects in IL-10 and its receptor genes IL10RA and IL10RB.80 ,81 Regulatory T cells, including natural regulatory T cells and Tr1 cells, but also macrophages and B cells, contribute to intestinal IL-10 secretion, and multiple cell types are IL-10 responsive. In a study analysing 66 patients with very early onset IBD, five patients carried mutations in the IL10RA gene, eight patients in the IL10RB gene and three patients in the IL-10 gene.82 Somewhat surprisingly, current data suggest a very similar phenotype in IL10R1 and IL10R2 deficient children,82 suggesting a dominant role for IL-10 in intestinal as well as in non-intestinal lesions and a limited function for the IL-22, IL-26 or INF-λ cytokines that share IL10RB but not IL10RA signalling with IL-10. Whereas conventional therapy, including exclusive enteral nutrition, steroids, immunomodulatory and anti-TNFα treatment as well as surgery, was not effective in treating children with IL-10 signalling defects in the past, haematopoietic stem cell transplantation has been successfully used to induce remission of IBD-like immunopathology.80–82
Genetic association studies suggest a role for IL-10 and IL10RB signalling in patients with IBD, in particular UC.3 ,83 The gut inflammatory phenotype in mouse models initiated several pilot studies to use the anti-inflammatory potential of IL-10 in patients with IBD. Initial trials using IL-10 were complicated by systemic side effects and the short half-life of the molecule but targeted and local application regimens might revive this potential treatment option in stratified patient subgroups with IBD.84
Defects with unknown colitogenic mechanisms
As attempted above, for a substantial proportion of immune defects a hypothetical colitogenic pathomechanism can be affiliated. For other genetic defects, the functional understanding is largely not clear. Thus loss of function mutations in DOCK8 cause autosomal recessive hyper-IgE syndrome and may predispose to colitis, but its functional role is currently not defined.85 Similarly, colitis has been described in several patients with trichohepatoenteric syndrome caused by TTC37 and SKIV2L defects.86 ,87 Both gene products are likely involved in the RNA exosome multiprotein complex that is involved in the decay pathways of normal mRNA and RNA surveillance processes.87 Whether a defect in epithelial stress response or immune dysfunction predispose to colitis in trichohepatoenteric syndrome needs to be shown.
IBD-like inflammation results from multiple layer mucosal defects and complex immune deviation
The above described simplified classification of defects with an IBD-like phenotype highlights the fact that most genetic defects affect multiple layers of intestinal barrier function and immune competence. Indeed, most diseases effect different cell types either because the gene is expressed by multiple cell types (such as WAS, PTEN, IL10RA, IL-10RB) or because the gene is expressed on a cell but might provide essential signals to interacting cells (such as ICOS, IL10). In addition, further cell types can be involved due to autoimmunity. Indeed, there are multiple defects that lead to autoimmune or haemophagocytosis induced neutropenia, and several immunodeficiencies not regarded as primary B cell defects are associated with low B cell numbers and/or immunoglobulins (SKIV2L, TTC37). Multiple disorders have a Th2 bias, as indicated by high IgE and/or eosinophilia in FOXP3, IL2RA, IKBKG, WAS or DOCK8 deficient patients. Classical phagocyte defects such as CGD or complex disorders (WAS, PTEN) may affect regulatory T cell function.
Variable penetrance of the IBD phenotype
The (nearly) complete penetrance in IL-10 signalling defects or IPEX syndrome together with a severe phenotype (ie, infant age onset of intestinal inflammation and lethal complications) suggests true monogenic IBD-like immunopathology in those two groups. The majority of the discussed defects however have a moderate or low penetrance of 2–30% for the IBD-like immunopathology, even if the disease defining phenotype (ie, immunodeficiency) is monogenic. Although a penetrance of 5% for the IBD phenotype is obviously low, compared with the strongest CD associated variants in NOD2, this association is still substantial. Indeed, recent data suggest that CD penetrance at the age of 50 years was 0.3% for heterozygotes and 1.5% for compound heterozygotes and homozygotes NOD2 variants.88 Larger studies on monogenic defects need to clarify the true penetrance for most defects as selection bias might affect penetrance data in opposite directions— that is, overreporting of rare variants with the IBD phenotype in the published literature as well as underreporting of rare variants in the IBD cohort.
Role of gene–environment and gene–gene interactions
A substantial rise in the incidence of IBD within the past century suggests the important influence of environmental factors for human intestinal inflammation.89 Most mouse models of intestinal inflammation highlight the essential role of the intestinal microbiota as development of colitis (even in mice deficient in essential immunoregulatory molecules such as IL-10) depends on the intestinal colonisation by individual microbes or bacterial communities. It is therefore attractive to speculate that the bacterial flora could play an essential role for intestinal inflammation in patients with monogenic defects but clear evidence for this is currently lacking and current therapeutic manipulations such as antibiotics or probiotics are not established or largely unsuccessful in this group.
In addition to environmental factors, common genetic variants may modify the phenotype in patients with an underlying rare genetic defect. A potentially pathogenic c-terminal XIAP mutation co-segregated with a CD40L polymorphism in one family with XLP-2, leading to a modified activation and apoptosis in T cells and B cells in vitro.90 Packwood et al investigated the effect of CD associated NOD2 mutations as modifier gene towards an enteropathic or granulomatous phenotype in 285 CVID patients. The NOD2 Gly908arg polymorphism was more prevalent in a subgroup of CVID patients with autoimmune disorders or enteropathy compared with controls.91 As this association was borderline significant, has not been replicated and CVID is a complex disorder, it will be interesting to revalidate those findings once such a cohort can be stratified according to the CVID causing mutational spectrum.
Related to the speculation that common genetic variants might influence the phenotype of rare orphan diseases, it will be interesting to see, how rare, possibly compound heterozygous or hypomorphic private mutations in the 40 (or more) rare disease genes with IBD-like phenotype influence the disease in IBD patients on a population level.
Rare variants causing IBD-like and IBD inflammation: role for next generation sequencing
As in other fields, next generation sequencing, including whole exome sequencing, has enabled to detect IBD-like causing rare genetic variants and to define new diseases.51 ,64 ,92
One of the lessons from these exome sequencing studies is that already known disease causing genes were found in patients with non-classical phenotypes. Phenotype–genotype predictions might be more accurate in the future but variable phenotype could also constitute a general problem to oversee and test the diversity of potentially colitogenic mutations. Given the increasing number of genes associated with IBD-like pathology and the reduced costs of sequencing, multiplex gene sequencing or whole exome sequencing platforms will increasingly be cost effective, instead of using gene by gene analysis, and will likely complement the current phenotyping by endoscopy and immunological follow-up (oxidative burst assay, immunoglobulins, lymphocyte subsets, etc).
Whole genome and whole exome sequencing as well as copy number analysis will increasingly allow to investigate a large number of rare heterozygote defects, such as modifier genes.93 Indeed, it is very likely that not only the known autosomal dominant loss of function (PTEN, RET) or gain of function mutations (GUCY2C, PLCG2) modify the risk of IBD-like phenotype but that a large group of genes with heterozygous mutations contribute via a gene dosis effects. Recently, heterozygous mutations in IL-10 or IL18RAP with predicted damaging effects have been reported in a small group of paediatric onset IBD.94 As this might be a chance finding in a small group of patients, larger groups, statistical linkage as well as functional studies will be needed to assess whether heterozygous rare variants in IBD candidate genes have a relevant functional impact.
How informative are ‘orphan’ diseases for the understanding of IBD?
It is interesting to speculate that the complexity and diversity found within the monogenic diseases can inform on the diverse mechanisms that predispose to IBD. Analysis of 163 key genetic loci associated with IBD predict a dendritic cell and T cell dominated immune response.3 The role of B cell responses and granulocytes is less clear according to this analysis but monogenic diseases would certainly highlight the important role of granulocytes and macrophages and to a lesser extent B cells. The disease pattern shown in monogenic diseases may furthermore helpful in the functional understanding of IBD phenotypes. Indeed, diseases with granulocyte/phagocyte defects, as well as hyperinflammatory macrophage activation disorders and IL-10 signalling defects develop a CD-like phenotype with granuloma formation and fistulising disease in a substantial number of patients. A stringent genotype–phenotype analysis of IBD patients will be needed to assess whether there are similar phenotype relations in the disease spectrum of patients with common IL-10 and IL10RB variants.3 ,83
In addition to phenotypic stratification, monogenic disorders might inform on novel therapies.
As subsets of patients with hyperinflammatory monogenic disorders, in particular mevalonate kinase deficiency, produce excess amount of IL-1β and treatment with IL-1β antagonists has therapeutic benefits in those patients,47 ,49 it is tempting to speculate whether subsets of patients with IBD and hyperinflammatory phenotype might similarly benefit from those treatments.
Other monogenic diseases remind us of the double edged sword of interfering with inflammatory signalling pathways. Largely due to its possible proinflammatory role in TNFα release, ADAM-17 has been proposed as a potential therapeutic target for inflammatory diseases, including IBD.95 Both human ADAM17 deficiency as well as animal data suggest that pharmacological blockade of this enzyme might lead to substantial intestinal side effects. Similarly, XIAP deficiency reminds us that XIAP inhibitors might have anti-inflammatory as well as colitogenic potential.96
Furthermore, haematopoietic stem cell transplantation has been successfully applied in several monogenic disorders. Pilot studies indicate a potential benefit for subgroups of patients with severe IBD (supplemental citation131). In light of the side effects of this treatment option, lessons from monogenic disorders and genetic stratification might become helpful for selection of IBD patients with a potential benefit and might help to exclude patients with increased risk but limited success due to epithelial dominant defects.
Conclusion
Understanding the pathogenesis of extremely rare monogenic diseases with high clinical impact can complement our understanding of complex multigenic intestinal inflammation in IBD. Monogenic diseases highlight multiple layers of mucosal barrier in the gastrointestinal immune system, including epithelial barrier, granulocyte function as well as hyper- and autoinflammatory macrophage activation, adaptive immune selection and negative immune regulation.
References
Footnotes
-
Correction notice This article has been corrected since being published. For consistency, in figure 4 CD25, IL10R1/IL10R2 has been exchanged by the gene name IL2RA, IL10RA and IL10RB.
-
Acknowledgements Many thanks to Kevin Maloy, Fiona Powrie, Chris Schiering, Bodo Grimbacher and Klaus Warnatz for comments. Due to space constrains and the broad field covered, I was not able to include all relevant references of all diseases and apologise to the omitted authors.
-
Competing interests Industrial project collaboration included UCB Pharma and Vertex Pharmaceuticals. Reagents were received from Adeypharm, Pfizer and Merck. Travel support was received from GSK foundation (2004), Essex Pharma (2010) and MSD (2012).
-
Provenance and peer review Commissioned; externally peer reviewed.