Article Text

Abstract
When growing up, the pharmacokinetic (PK) and pharmacodynamic (PD) profiles of drugs change, which may alter the effect of drugs. To ensure optimal drug efficacy and safety in paediatric care, PK and PD relationships of drugs need to be explored in children. This article presents an outline on performing a population PK/PD study and translating these results into rational dosing regimens, with the development and prospective evaluation of PK/PD derived evidence-based dosing regimen being discussed. Examples on amikacin, morphine and busulfan are provided, showing how PK(/PD) modelling not only led to optimization and individualization in paediatric clinical care for the specific drugs but also to insight in maturation of organ systems involved. It is shown that the latter results can subsequently be used as a basis for dosing of other drugs eliminated through the same pathway. Ultimately, these efforts should lead to predictable drug efficacy and safety across all age groups.
- PKPD
- PK/PD
- child
- pediatrics
- population modeling
Statistics from Altmetric.com
Introduction
Many drugs used in daily paediatric practice lack an evidence-based dosing regimen. A recent review shows 13–30% of drugs used in primary care setting and 49–87% in hospitals are prescribed in an off-label or unlicensed manner.1 Off-label doses in children are often empirical, based on body weight in a linear manner, and derived from an extrapolated adult dose. Using per kg doses, one assumes that the dose to achieve comparable concentrations increases in a linear fashion with weight. In addition, the assumption is often made that children and adults have a comparable concentration-response relationship. Since developmental changes are mostly non-linear, empirical dosing can lead to overdosing or underdosing, especially in specific age groups such as neonates, in particular extreme low birthweight infants, thereby possibly introducing toxicity or reduced efficacy.2
In order to prescribe drugs in children in an evidence-based manner a thorough understanding of the pharmacological profile in children is needed, since the response to drugs may vary highly between children and adults. When growing up, among others, body composition changes, enzyme pathways and renal function mature thereby influencing the pharmacokinetics (PK) of drugs. Maturation also occurs in the expression and function of proteins and receptors, which may alter the effect of drugs (pharmacodynamics (PD)). Also, maturation rates are known to vary between organs and within organs between metabolic pathways.3 To describe these processes, the PK as well as the PD need to be investigated in a wide age range.4 ,5 Subsequently, evidence-based dosing regimens can be derived.6–8 The so-called population approach has highly facilitated PK/PD modelling in children because it enables the analysis of sparse sampling datasets and/or datasets derived from clinical practice in which different doses have been applied.9–11
This article presents an outline how to perform a population PK/PD study and how to translate these results into evidence-based dosing regimens. The approach to reach individualised dosing guidelines in children based on population PK/PD modelling is explained, after which examples are presented and clinical implications as well as perspectives of this approach are discussed.
PK/PD modelling on the basis of a population approach
The concentration-time profile of a drug in blood is determined by several processes such as absorption, distribution, metabolism and elimination, and the parameters characterising these processes such as clearance and bioavailability can be calculated from this profile. Since individuals show variability in concentration-time profiles and thus PK parameters, concentration-time profiles of different individuals are needed, resulting in a mean value for each parameter with a distribution.
If all parameters were to be estimated in each patient separately, a substantial number of samples per patient are required in order to describe the entire profile. This obviously is not an ethically justifiable approach in paediatric and in particular neonatal medicine. Also, the dosing regimen and the number of samples have to be roughly the same to allow for comparison between patients. Another disadvantage of this approach is the inability to distinguish between-subject variability (BSV) and error, a variable containing information on measurement errors, wrong notation of sample times and model misspecification. This may result in overprediction of BSV, leading to large confidence intervals of parameters.
Nowadays, using advanced software in combination with high computing power, the population approach is the preferred method of PK/PD analysis.12 Using this method, instead of estimating parameters individually followed by a statistical analysis, all available data from all individuals are pooled to estimate a population mean for all parameters. Subsequently, based on individual concentrations, the BSV and error are estimated separately. Here, BSV is a percentage indicating the difference between population mean and the individual value for each parameter. Since the whole dataset is used to estimate the population means, sparse and unbalanced (unequal distribution of samples over time and/or per patient, as is often the case) sampling can be done.
In summary, population PK/PD is the preferred choice, for ethical and practical reasons, as well as the more accurate estimation of model parameters.9–11
Designing an individualised dosing regimen
When performing a PK/PD study, drug concentrations and outcome/effect data are needed from each patient. Besides drug concentrations and effects, factors potentially influencing the PK or PD should be collected. These so-called covariates are important variables determining part of the BSV, and a crucial part of the individualised dosing regimen. Covariates may include patient related (weight, age, creatin clearance), disease related (severity, progression, duration) and/or treatment related (route of administration, comedication) factors.
Development of an evidence-based individual dosing regimen through population PK/PD modelling is optimally achieved using a multistep approach (figure 1).4 This approach consists of the following steps:
-
Optimal study design based on preliminary data
-
Development and internal validation of PK/PD model
-
External validation of the PK/PD model
-
Prospective validation in clinical study
-
Proposed individualised dosing regimen

Proposed multistep approach for modelling and simulation using non-linear mixed effects modelling for the optimisation of drug dosing in children. The four steps that are proposed are (1) optimisation of clinical trial designs based on simulations using preliminary data; (2) development and internal validation of population PK-PD models using sparse data; (3) external validation of the population PK-PD models using independent data; and (4) prospective clinical evaluation of the PK-PD model-based dosing regimen. PK, pharmacokinetics; PD, pharmacodynamics. Printed with permission from: Ince I et al. Drug Discovery Today 2009;14:316–20.
In the first step, based on literature or previous experience, a study has to be designed, optimising the number of patients13 and timing of the sampling windows. Since population PK/PD modelling can be done on unbalanced and sparse data, the number of samples per patient is of less importance. Bearing in mind that all samples of all individuals are analysed simultaneously, it can be anticipated that when all samples are taken on the same preset times after dosing, crucial information may be lacking between these concentrations thereby complicating the analysis.14
Second, with data generated from the performed study, a population PK model is developed, starting with the identification of a structural model. This comprehends a model adequately describing the data, typically not including covariates. Then a statistical model characterising BSV is selected, after which covariates are tested and selected. For example, in most paediatric studies, weight is a covariate for clearance and/or volume of distribution. The selected covariates need to be clinically relevant and feasible. The ultimate goal of the model is not only describing PK and PD in the population used to develop the model, predicting dosing in future patients is of much greater importance. In these patients, dosing can be adjusted based on the value of the included covariates.11 The above steps will be repeated for the PD model to describe the relationship between drug concentrations and effects.
During and after developing a PK/PD model, the different aspects of the performance of a model have to be tested by an internal validation procedure. Validation procedures are of particular relevance when sparse datasets are analysed.15 For example, predictions need to be accurate in low and high concentration ranges and in different weight ranges despite the limited number of observations. Predictions of concentrations are tested using the diagnostic plots, where predictions are plotted against measured concentrations. On the other hand, to test for subgroups which might influence the performance, internal validation can be performed using resampling and predictive techniques.15–17
As a third step, external validation is executed using another dataset when available. Here, the developed model is evaluated using another dataset, the performance of the model is checked and parameter values are compared with the values of the original dataset. The external dataset should be comparable to the original in terms of the included covariates as extrapolations may introduce bias.
With the model being internally and externally validated, a new dosing regimen will be proposed based on the model. In this fourth step, data simulation is a helpful tool. Here, the different parameters (ie, clearance, volume of distribution) can be fixed to their estimated value. Now, when varying the dose, blood concentrations over time can be simulated, making it possible to select the optimal dosing regimen for each individual child with a given body weight and age.
In a subsequent prospective study performed as a proof of concept, the developed individualised dosing regimen is evaluated. The goal of this evaluation is to verify whether predicted outcomes match observed outcomes in terms of PK (exposure) and/or PD (drug effects) across all age groups studied.
Examples of population PK-derived evidence-based dosing regimens
Amikacin
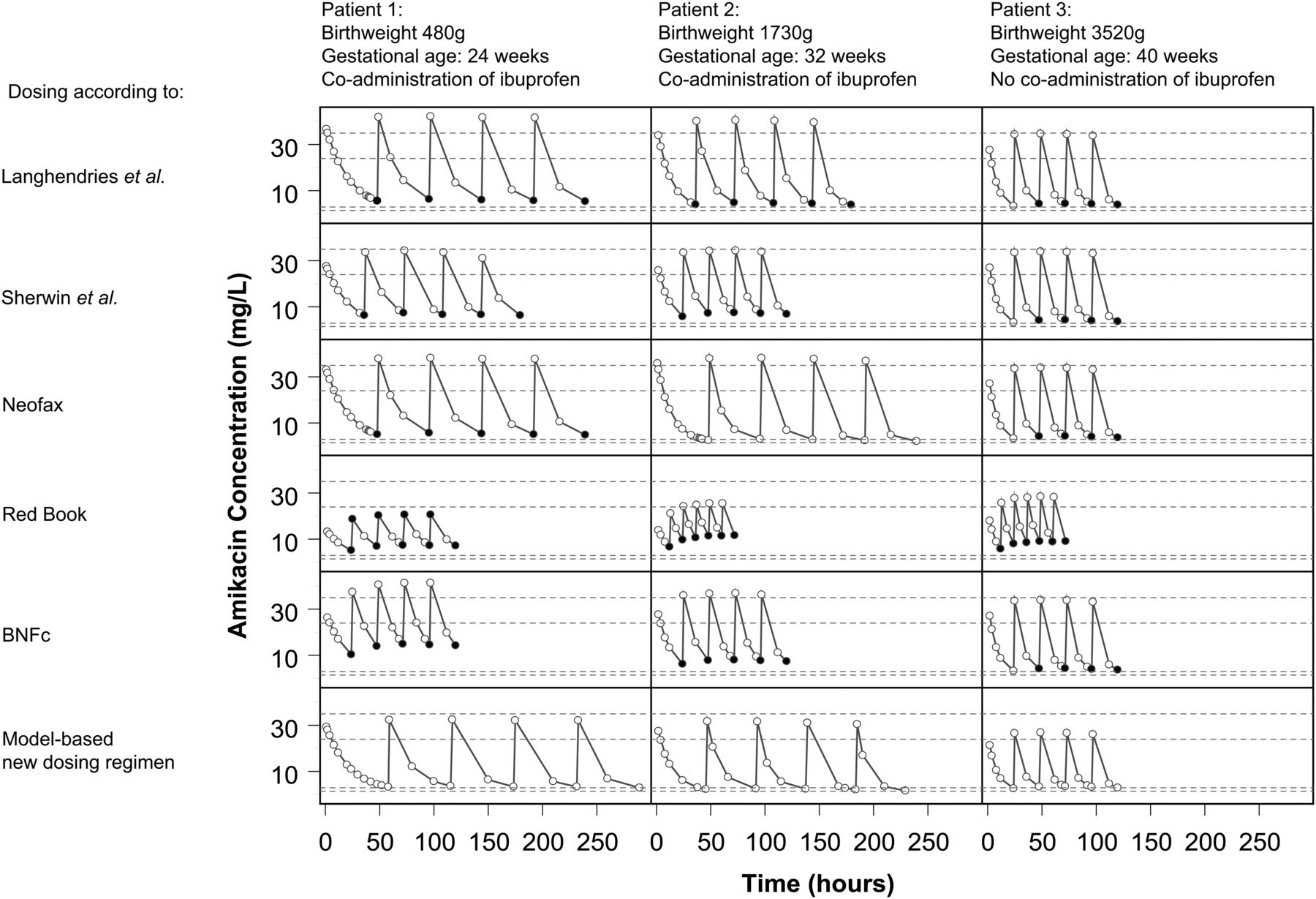
An example in which population PK-modelling has lead to an individualised dosing regimen is a study on the PK of amikacin, which is an antibiotic drug that is almost entirely eliminated through glomerular filtration, in a large dataset of more than 800 preterm and term neonates.18 Amikacin clearance proved related to postnatal age and birth weight, that is, children with higher age and weight having a faster maturation of clearance. Another covariate included in this model was the coadministration of ibuprofen, which reduces amikacin clearance. With this model, an evidence-based dosing regimen was proposed by performing simulations with the developed model. In contrast with most current paediatric dosing regimens, the proposed dosing regimen uses birth weight in combination with postnatal age to calculate the appropriate dose instead of weight.18 When comparing simulated concentrations both of the new regimen as well as various current dosing regimens, including the Red Book,19 the proposed evidence-based model was shown to be superior to all other regimens in terms of achievement of target peak and trough concentrations (figure 2). Thus, in contrast to established dosing regimens, the model-based individualised dosing regimen may be anticipated to prevent toxicity while remaining efficacious.18 Currently, the results of a prospective study in more than 600 neonates in which the individualised dosing regimen18 is evaluated are being analysed.

Model-based predicted concentration-time profiles of amikacin for three typical neonates, using five different currently used dosing guidelines19 ,34–37 and according to the new model-based dosing regimen. Open dots: concentrations within target range, black filled dots: Cmax over target Cmax, grey filled dots: Cmin under target Cmin. Cmax=maximum concentration; Cmin═minimum concentration. Long dash line, Limit of target Cmax range; Short dash line, limit of target Ctrough range. Adapted from De Cock RFW et al. Maturation of the glomerular filtration rate in neonates, as reflected by amikacin clearance. Reproduced from Cock R. F. W. De et al. Maturation of the glomerular filtration rate in neonates, as reflected by amikacin clearance. Clinical pharmacokinetics 2012;51:105–17 with permission from Adis (© Springer International Publishing AG 2012. All rights reserved.)
Morphine
Morphine is a commonly used opioid in children for which the feasibility of PK/PD-models to derive evidence-based dosing regimens was shown.9 Modelling of morphine in children of 0–3 years old including premature neonates showed the increase of clearance through uridine glucuronosyltransferase (glucuronidation) to be higher in older children than in neonates.20 In addition, neonates younger than 10 days proved to have a 50% lower glucuronidation clearance. Using the non-linear dosing regimen based on this model, a narrow range of serum concentrations of morphine can be achieved in this age group, despite a broad variation in clearance20 (figure 3). This model was externally validated using six datasets which were not used for the development of the model.21 Using this model, an evidence-based dosing regimen was developed, which was prospectively validated in a double-blinded clinical controlled trial (table 1).22 In this clinical study, patients postoperatively received a morphine loading dose followed by either paracetamol or morphine infusion. In case of pain, morphine rescue doses were given in both treatment arms. Morphine dosing in both the morphine treatment arm as well as rescue medication were given according to the individual dosing regimen, with children younger than 10 days receiving a 50–75% lower dose compared with current standards, while older children received a higher dose. Using this dose, efficacy was maintained in the majority of children, while the risk of overdosing was reduced, especially in young neonates. However, although blood concentrations were comparable, rescue doses were frequently needed in the relatively older children, suggesting a difference in PD relation in different age groups. This may be caused by a difference in sensitivity to morphine and its active metabolites or a difference in distribution to the target sites. To date, the morphine target concentration to achieve adequate pain relief is unknown and may vary between children in different age groups. Incorporating this PK-PD relationship and the effect of age on this may improve the proposed model and thus the dosing regimen.
Dosing table for individualised maintenance dose for children based on the developed evidence-based dosing regimen and the dosing based on the current regimen

Simulation of morphine concentrations in children weighing 0.5 kg, 1 kg, 2 kg, 2.5 kg and 4 kg with a postnatal age <10 days (dotted lines) and children weighing 0.5 kg, 1 kg, 2 kg, 2.5 kg, 4 kg, 10 kg and 17 kg with a postnatal age of >10 days (solid lines) based on a dosing scheme containing a loading dose of 100μg/kg followed by a maintenance of 10 μg/kg/hr (A) or 10 μg/kg1.5/hr with a 50% dose reduction in children <10 days old (B). Adapted from Knibbe CAJ et al. (2009). Morphine glucuronidation in preterm neonates, infants and children younger than 3 years. Reproduced from Knibbe, CAJ et al. (2009). Morphine glucuronidation in preterm neonates, infants and children younger than 3 years. Clinical pharmacokinetics 2009;48:371–85 with permission from Adis (© Springer International Publishing AG 2012. All rights reserved.)
Busulfan
A third example is the development of an individualised dosing regimen used in paediatric haematopoietic cell transplantation (HCT). It has been shown before that individualisation of dosing of the various drugs used in this procedure, including chemotherapy, can improve morbidity and mortality.23–25
Busulfan is one of the chemotherapeutic drugs used in paediatric HCT as preparative chemotherapy. This drug has a narrow therapeutic window, with underdosing as well as overdosing leading to significant morbidity and mortality. An international cohort of 245 patients receiving busulfan in HCT was included in a retrospective PK/PD study.23 Ages varied from 1 month to 26 years, and patients received HCT for various indications. In this model, body weight proved an important factor influencing volume of distribution and clearance (figure 4). The dosing regimen based on this model is expected to lead to an optimised exposure in all body weight ranges when compared with the approved dosing, because it is aiming for an AUC that previously has shown to result in the highest event-free survival.26 After external validation of this model,27 the dosing regimen is now used for dosing busulfan in current clinical practice (table 2).
The model-based individualised dosing table for busulfan, aiming for a myeloablative (AUCday0–4 of 90 mg*h/L in combination with fludarabine), compared with the approved dose in the summary of product characteristics (SPC)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Individual predicted (post hocs, presented as dots) and population predicted (lines) values for busulfan clearance versus body weight. Data is presented on a log scale and on a normal scale (insert). Printed with permission from Bartelink IH, van Kesteren C, Boelens JJ, et al. Predictive performance of a busulfan pharmacokinetic model in children and young adults. Therapeutic Drug Monitoring 2012;34:574–83.
Discussion and perspectives
In children, with their changing body composition and maturation in function of metabolising and/or eliminating organs and receptors, evidence-based dosing regimens are crucial. With empirical dosing, which is used in a substantial portion of drugs prescribed in paediatric practice, drug effects may vary over age and weight. This may potentially lead to a decrease in drug effect or toxic doses with an increase in side effects in one or more specific age groups.
Population PK/PD-modelling is a validated tool for developing evidence-based dosing schemes.8 ,10 Using this technique, provided a proper internal validation procedure is performed, a model can be built based on sparse and unbalanced data, which is common in paediatric practice due to practical and ethical restrictions of frequent blood sampling.11 ,12 With this model, the relationship between exposure and effects of drugs can be precisely predicted to ensure a constant dose-effect relation in a population, including children in varying age groups including neonates.9
When conducting a study to develop a dosing regimen through PK/PD, the multistep approach (figure 1) is the preferred choice.4 Using this approach, an informative database is derived on which a PK/PD model can be built after which model performance is extensively validated. Nonetheless, a robust PK/PD model can still be built with limited information. This approach has proven its value in past studies resulting in solid evidence-based dosing regimens.18 ,20 ,23 It should be noted that, when completing the steps in this approach, there may still be room for optimisation of the model and the proposed dosing regimen. With increasing amounts of data, special populations (ie, renal impairment, liver disease) and other conditions resulting in outliers can be identified, after which dose corrections for these groups can be implemented in the regimen.
Besides the development of individualised dosing regimens, a validated PK model may also be used to understand metabolic pathways. Acquired knowledge on a biological system on the basis of a paradigm drug can be used to translate an existing model to other drugs that use the same metabolic or elimination pathway.9 ,10 System-specific parameters (maturation of clearance as quantified in the identified covariate model) can be obtained from previous work on paradigm drugs, so that only drug-specific parameters (population value of volume of distribution or clearance) have to be estimated, which drastically reduces the number of patients and samples needed.9 ,10 Interaction with Physiologically Pased PK (PBPK) modelling groups28 is of great value in this context which has resulted in successful predictions of weight-related changes in glucuronidation clearance of zidovudine and potentially many other uridine glucuronosyltransferase substrates on the basis of the modelling results in morphine.29 ,30 Also, the amikacin model characterising weight and age-related changes in amikacin clearance in preterm and term neonates18 was able to precisely reflect maturation of glomerular filtration and thus predict the dosage regimens of other renally excreted drugs by glomerular filtration in neonates.31 ,32 These examples demonstrate how the development of evidence-based dosing regimens can be accelerated in a sophisticated manner.
However, it should be noted that, although the development of evidence-based dosing regimens using PK models is often a major improvement compared with empirical dosing, more emphasis should be put on the characterisation of the PD relation in children.5 It is both the dose-concentration and concentration-effect relationship across the paediatric age range that should be explored in order to achieve predictable efficacy and safety in all individuals, as well as the effect of covariates such as age on these relationships, which may lead to further improvement of dosing regimens. In the examples presented in this paper, surrogate PD end points (ie, target peak and trough concentrations associated with efficacy and toxicity, respectively) were available for amikacin18 and target AUCs associated with optimised efficacy with acceptable toxicity were available for busulfan.26 Further research should therefore focus on PD relations in children, with the field of pain research as an example where many efforts have been put on the validation and use of age-related PD end points that may serve as a basis for the PK/PD studies in children.33
In the future, evidence-based derived dosing regimens in paediatrics need to become the norm rather than the exception. In order to achieve this, paediatric PK/PD studies need to be conducted in both the development of new drugs and in drugs that have been on the market for a short time or a long time. The examples shown here demonstrate how PK (/PD) modelling may lead to optimisation and individualisation in paediatric clinical care for the specific drugs but also for other drugs eliminated through the same pathway. While this could potentially accelerate the development of dosing guidelines for many drugs, ultimately, these and other efforts should lead to predictable drug efficacy and safety across all age groups.
References
Footnotes
-
Contributors Concept, design, drafting of paper: RA, CvK, CAJK. Critical revision: JJB, RGMB, DT, CAJK.
-
Funding The work of CAJK is supported by the Innovational Research Incentives Scheme (Vidi grant, June 2013) of the Dutch Organisation for Scientific Research (NWO). The work of RA and CvK was supported by ZonMW grant number 40-41500-98-11044.
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.