Article Text

Abstract
Objective A genetic opinion is frequently requested in the assessment of a child with suspected fetal alcohol spectrum disorders (FASD). We studied the outcome of genetic assessment of 80 children referred to a regional genetics centre between 2004 and 2010 to identify the value of the genetic assessment in cases of suspected FASD.
Design Retrospective case series.
Patients 80 patients, aged between 1 month and 26 years.
Methods Data from the medical records was abstracted, entered onto a standard study pro forma, recorded in an Excel spreadsheet and analysed using simple frequency analysis.
Results In 20% of cases fetal alcohol syndrome was confirmed at the genetic consultation. The most common facial features were thin upper lip (86.6%) and short palpebral fissures (82%). A lip–philtrum score of 4 or 5 was identified in two-thirds of cases. The most common alternative diagnosis was a chromosome disorder, representing 8.75% of the FASD referrals.
Setting A regional genetics service in the North West of England.
Conclusions Genetic assessment was of particular value in excluding other diagnoses and providing information to carers. Two-thirds of the children referred were subject to a care order increasing the difficulty to obtain a family and alcohol exposure history. Classification of FASD was difficult in children under a year old when data on growth and development were limited. Structural malformations were not common in the group overall and some previously reported diagnostic signs were not found to be reliable markers of FASD. Chromosome disorders showed phenotypic overlap with FASD and are an important differential diagnosis.
Statistics from Altmetric.com
Introduction
In 1973, a landmark report on birth defects in children of alcoholic mothers was published,1 which correlated prenatal alcohol exposure with a pattern of major and minor malformations, growth failure and cognitive and behavioural problems. Though this condition was initially designated ‘fetal alcohol syndrome’ (FAS), the term fetal alcohol spectrum disorders (FASD) has been proposed more recently to describe a broader range of adverse outcomes of prenatal alcohol exposure. The incidence of FASD is thought to range from 0.2 to 2.0 per 1000 live births.2 Diagnostic criteria for FASD were defined in 1996 by Stratton et al,3 and five diagnostic categories were proposed; FAS with confirmed maternal alcohol exposure, FAS without confirmed maternal alcohol exposure, partial FAS with confirmed alcohol exposure, alcohol related birth defects (ARBD) and alcohol related neurodevelopmental disorder (ARND). These criteria were subsequently updated by Hoyme et al in 20054 and further comprehensive guidelines have been published subsequently.5 ,6 Despite this guidance and the introduction of more objective scoring systems for FASD,7 it can still be difficult to confirm or refute this diagnosis in an individual child. Biomarkers for prenatal alcohol exposure exist, most notably the levels of fatty acid ethyl esters in meconium and hair,8 ,9 but utilising these in routine clinical practice has proven difficult. Obtaining an accurate history of alcohol exposure can be problematic as mothers may not be willing to disclose their true alcohol intake during pregnancy.10 A number of prenatal screening tools exist for identification of mothers at an increased risk of having a child with FASD including T-ACE, TWEAK and AUDIT-C.11 Many children presenting for diagnosis of FASD do not live with their mothers, making the use of these tools difficult.
What is already known on this topic?
▶ Exposure to alcohol in pregnancy can harm the unborn fetus.
▶ The effects of prenatal alcohol exposure vary depending upon the timing of exposure and blood alcohol concentration.
▶ There is no biomarker for FASD; this is a diagnosis of exclusion based on medical and alcohol exposure history together with suggestive clinical signs.
What this study adds
▶ The most important role of the clinical geneticist in the assessment of children with suspected FASD is to rule out alternative or co-existing diagnoses.
▶ Chromosomal imbalances may have phenotypes which overlap with FASD and microarray analysis should be considered when family history or clinical signs are suggestive.
▶ Obtaining an accurate history of prenatal exposure to alcohol is regarded as crucial for arriving at a diagnosis.
BMA guidelines recommend referral of all children with suspected FASD to a clinical geneticist.12 A genetic assessment is similar to other consultations involving the processes of history taking, clinical examination and investigation and tying all these together to arrive at a diagnosis. However, more attention is paid to the family and perinatal history and a detailed examination for morphological differences is carried out. The geneticist may order specific investigations such as chromosomal microarray analysis and single gene tests. When searching for a diagnosis, the clinical geneticist uses a wide variety of resources including specialised literature, databases and opinions from other colleagues. Some clinical geneticists question the value of their input to the diagnosis of FASD, which is an environmental disorder where there is often limited maternal and family information available. We therefore sought to explore the value of genetic assessment of children with suspected FASD in the setting of a regional genetics centre.
Methods
Patients
All patients registered as having a referral diagnosis of possible FASD who had attended the Manchester Genetics Service between January 2003 and January 2010 were surveyed. The medical records were identified and data abstracted from these. Each individual had undergone a standard genetic assessment with every case having been reviewed subsequently by at least one consultant clinical geneticist before deciding on the final clinical diagnosis. Information pertaining to age at the appointment, gender, family history, alcohol exposure history, pregnancy and birth, medical history, growth parameters and presence of major and minor malformations was gathered, together with the results of any investigations, differential diagnoses considered and the final clinical diagnosis. Information on individuals accompanying the child to the clinic appointment and whether the child was fostered, adopted or with birth parents was also collected. Clinical photographs were reviewed jointly by the authors and examined for facial features which are characteristic of FASD, in particular the lip–philtrum score.13 ,14 Individual features were itemised and checked for correlation with those reported in medical records. Finally, a judgment was made in each case as to whether the genetic assessment had contributed to the medical or other management of the individual seen. The criteria for this are outlined in table 1.
Contribution to medical management of patients.
Study population
A group of 80 patients, aged 1 month to 26 years fulfilling the above criteria was identified. This did not represent complete ascertainment of all possible FASD patients during this period as it did not include patients diagnosed with FASD with a different referral diagnosis. The latter group were omitted as they were difficult to ascertain completely from departmental databases and partial ascertainment based on clinician recall could have led to ascertainment bias. Individuals were classified into eight groups based on the final diagnosis. These were the four acknowledged categories of FASD (FAS, Partial FAS, ARND, ARBD) and four further groups which were ‘Indeterminate diagnosis’, ‘Normal child’, ‘Other syndrome’ and ‘Effects attributable to environmental neglect’.Photographs of 52/80 patients were available for assessment of FASD features.
Data collection and analysis
All data were entered onto a standard study pro forma and then inputted to an Excel spreadsheet. The data were analysed using simple frequency analysis to identify common findings across the whole group and significant differences between diagnostic groups.
Results
Description of cohort
The baseline characteristics of the cohort of 80 patients are shown in table 2. The mean age at referral was 5.5 years and 43% of patients were referred between 1 and 5 years. A total of 32 children (39.5%) were referred from a community paediatrician.
Baseline characteristics of a cohort of 80 patients presenting to a clinical genetic service with suspected fetal alcohol syndrome
In all, 59 children (73.75%) were either subject to a care order at the time of referral or attended with adoptive parents. Biological parents attended the session in 16 cases (20.25%). A social worker was present at the appointment in nine (11.40%) cases.
An adequate nuclear family history, defined as having obtained medical and developmental information about parents and siblings, was obtained prior to or at the time of the consultation for 55 referrals (68.75%). An incomplete family history was obtained in a further 14 cases. Information about grandparents and other individuals in the extended pedigree was not usually available. Of note, in 42% (29/69) families, there was a family history of learning disability.
An adequate history of alcohol exposure, defined as establishing with certainty whether alcohol had been consumed during the pregnancy, was obtained for 42 referrals (52.5%). In only 25 (31.25%) of these cases was an accurate assessment of the units of alcohol consumed per week possible. In just over half of all cases (n=41), there was reliable evidence that the mother had also taken recreational drugs during the pregnancy.
Genetic testing was carried out in 62 patients. Of these, routine karyotype was checked in 48, 22q11 fluorescence in situ hybridisation in 23 and Fragile X in 21. Ten patients had chromosomal microarray testing. In two cases, no testing was undertaken because consent was not available and testing was not requested in 10 cases.
Overall, 16 (20%) of the patients received a final diagnosis of FAS with or without confirmed maternal alcohol exposure, 15 (18.75%) were labelled as having PFAS, six had ARND (7.5%) and five ARBD (6.25%). Twenty-six cases were designated as ‘indeterminate’. In seven cases (8.75%) an alternative syndromic diagnosis was made, six of which were chromosomal microdeletion/duplication syndromes (table 3). In two cases, the child was deemed to be a normal child and in the remaining two cases the symptoms were considered to be most likely due to poor environment (figure 1).

Distribution of FAS referrals into diagnostic groups after the clinical genetic assessment. ARBD, alcohol related birth defects; ARND, alcohol related neurodevelopmental disorder; PFAS, partial fetal alcohol syndrome; FAS, fetal alcohol syndrome.
Differential diagnosis of this FAS series made on genetic testing
The characteristics of the children diagnosed with FAS or FASD were compared with those who were not diagnosed with FASD to define the main differences among the three groups (table 4).
Comparison of the characteristics of this FASD and non-FASD series
Children with a final diagnosis of FAS with or without confirmed maternal alcohol exposure
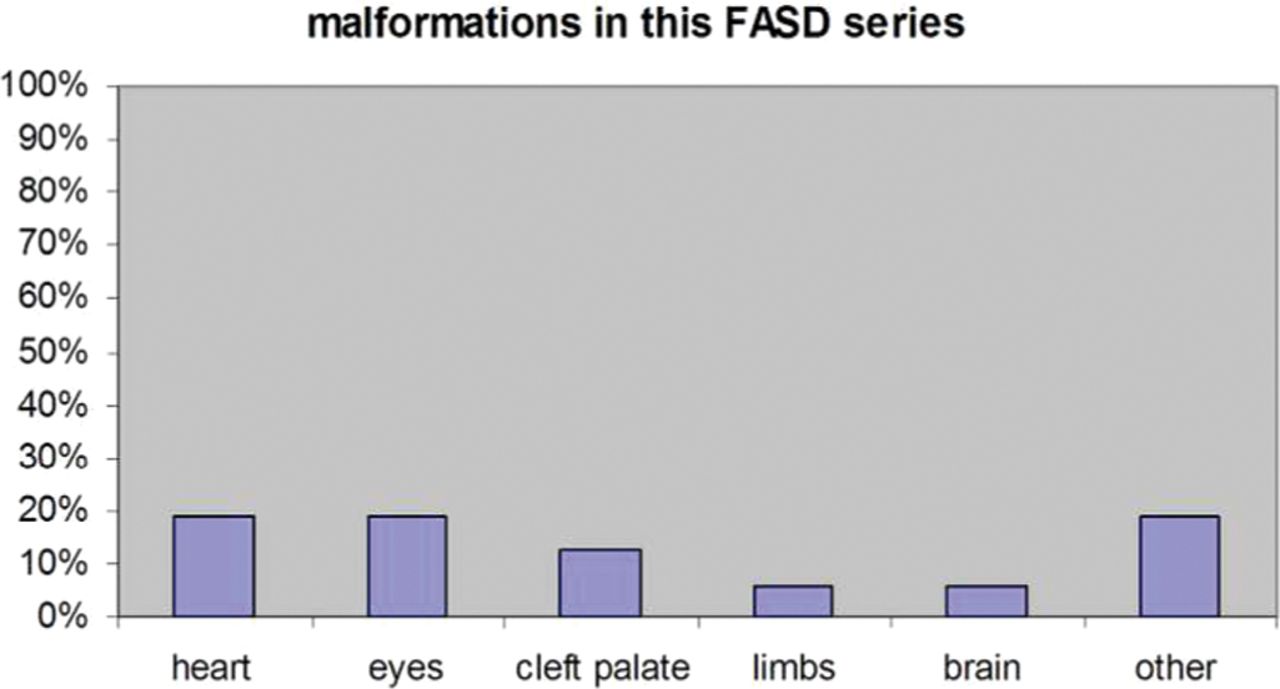
In 16/80 cases, the initial suspected diagnosis of FAS was confirmed at the genetic consultation (table 4). Of these, the history of alcohol exposure was definite in 56.25% and probable in 43.75%. The number of units of alcohol/week, when known, was highly variable (18–157). Fetal abnormalities were seen in 63.6% of the FAS cases on antenatal scans with intrauterine growth restriction (IUGR), seen in 50%, being the main abnormality. The main presenting features were intellectual disability, behaviour problems, growth failure, microcephaly and speech difficulties. The most common facial features were thin upper lip (86.6%) and short palpebral fissures (82%). A lip–philtrum score of 4 or 5 was identified in two-thirds of cases. Epicanthic folds, clinodactyly, anteverted nares and nail hypoplasia were less frequent. We identified ‘hockey stick creases’ in three patients diagnosed with FAS while none had a ‘railroad-track ear’. Associated malformations were present in fewer than 20% of cases. (figure 2).
{kind=link}
{kind=link}
Prevalence of malformations in this fetal alcohol spectrum disorder (FASD) series.
Children with an alternative diagnosis
The most common alternative diagnosis was a chromosome disorder. Abnormalities of 22q11 were identified in three patients after investigation by fluorescence in situ hybridisation. Two patients from the same family had a 22q11 duplication and one had a 22q11 deletion.
In nine cases, chromosomal microarray analysis was requested to screen for submicroscopic chromosome imbalances. This investigation was prompted if an individual had dysmorphic features out with the FASD spectrum or if there was a family history of learning disability or structural malformations.15 Imbalances were detected in four cases with one patient each having a 2p16.1p15 microdeletion, a 15q13.3 microdeletion and a 1q21 microduplication, and one patient having both 14q21.1 and 16p13.3 microduplications.
Patients with chromosome abnormalities represented 8.75% of the FASD referrals (7/80) with six of the seven chromosomal abnormalities being considered pathogenic (table 3). All of these had been described previously in the medical literature. There was a definite history of prenatal alcohol exposure in two children who also had an alternative diagnosis and a probable history in six. They had all presented with intellectual disability and behaviour problems. In more than half the cases, thin upper lip and short palpebral fissures were noted. There were associated malformations in all these patients, namely, three had a genitourinary malformation, a duplex kidney, hypospadias or hydronephrosis; two had capillary malformations; one presented with optic nerve hypoplasia; one more had a cardiac valve dysfunction; and the patient with the 22q11 microdeletion had palatal insufficiency. Prenatal alcohol exposure could also have contributed to the phenotype in these patients.
Impact on management
The clinical genetic consultation was judged to have impacted on the medical management of the child in 20/80 cases. This was either through diagnosis of another condition which required medical care or because the genetic opinion prompted screening tests for complications. Those judged to have benefited most from the genetic consultation were carers, usually either foster parents or adoptive parents, of whom 76% were provided with specific written information about the condition and/or contact information for support groups or services.
One of the increasingly common reasons for referral for a genetic opinion was so that the child could be assessed to assist with an adoption placement. It was not possible to determine whether the genetic assessment had facilitated this as the outcomes of the adoption process were not known.
Discussion
Main findings
The majority of children referred for diagnosis of suspected FASD were in or had been in the care system. This caused problems with obtaining details of the family history and alcohol consumption, both crucial points when making a diagnosis of FASD and excluding other diagnoses. A biological parent attended in only a fifth of cases and it was often extremely difficult to obtain detailed information on early health and development. Some information was made available from social work records which were often detailed but a social worker attended only 11.4% of consultations and often did not have this information to hand or had not been involved with that particular child's care before.
In 21% of cases the referral concerned a child under a year old, as efforts to organise early adoption placements are encouraged. In these cases, diagnosis was difficult since cardinal features of the FASD, such as developmental delay and behaviour problems, could not be evaluated at this age. Overall, the mean age at diagnosis of FASD in this series was 5.5 years. Where children were referred at an older age, this was often because behavioural problems had become apparent, prompting consideration particularly of ARND. Individuals aged 6–26 years old were less typically growth retarded at presentation compared with children under five, suggesting either that catch-up growth occurs in later childhood in FASD or confirming that the group which presents later is more likely to have ARBD or ARND which are not typically associated with growth impairment. Mean head circumference percentile in individuals presenting at 6–26 was smaller than that in the 0–5 group. This may indicate that in ARBD and ARND, alcohol is preferentially affecting brain growth rather than general growth. Philtrum scores did not change significantly among the two groups (table 5). Spohr et al16 reported a 20-year follow-up of 52 individuals with FASD and like us showed that microcephaly and some facial characteristics, such as a smooth philtrum, persist over time. We therefore suggest that microcephaly and a significantly high lip–philtrum score are useful indicators of FASD in the older individual. Spohr et al found significant differences in the height and weight of older individuals with FASD. Older male subjects tended to have short stature and be underweight whereas older female subjects tended to become overweight. In our study, mean weight and height were higher in older individuals but we were not able to compare these between sexes. Intellectual disability was one of the commonest presenting features and was seen more consistently in the group of children who received a final diagnosis of FAS or FASD. Where children had intellectual disability this had usually been formally assessed prior to referral. A variety of cognitive deficits may be seen in FASD, not just impairment of IQ,17 and geneticists referred to a number of professionals including community paediatricians, psychologists and psychiatrists where specific problems were suspected.
Age-related presentation characteristics of this FASD series
This study highlighted that the most useful role of the geneticist was to identify and investigate alternative diagnoses. Ultimately, 52.5% of referrals were confirmed as having FASD but a number of other differential diagnoses were considered including Fanconi anaemia, Smith–Lemli–Opitz syndrome, Stickler syndrome, Turner syndrome and Kabuki syndrome, all of which have features which overlap with FASD. Importantly, genetic testing carried out at the time of consultation identified the underlying diagnosis in 10% of referrals. Chromosomal microarray analysis, a new genetic technology which allows detection of much smaller chromosome imbalances than routine karyotyping was carried out only in nine selected cases having become available only towards the end of the study period. There were findings of clinical significance in three cases. We propose that microarray analysis should be considered in children with an FASD-like phenotype if there are atypical features or a family history of learning difficulties, structural anomalies or recurrent pregnancy loss. Although obtaining parental samples to interpret microarray results might be problematic, the finding of known recurrent rearrangements in six of our patients (including the 22q11 abnormalities which would have been detected on microarray analysis) provided a diagnosis without the need for parental chromosome examination. The 22q11 deletions occur at a frequency of around 1 in 8000. The classical symptoms of cleft palate, cardiac defects, speech difficulties due to velopharyngeal insufficiency and impaired growth and development overlap significantly with those of FAS.
Identification of chromosome imbalance may have significant implications for management and counselling of siblings and family members. Many chromosomal microdeletions have been linked to behavioural or psychiatric findings and one might hypothesise that the presence of such an imbalance, if present in the mother, could even have played a role in alcohol dependency.
Structural malformations did not seem to be a common finding in our cohort of FASD children, being present in a minority (18.75%) of cases. All patients with a chromosomal imbalance presented with a malformation, however.
A flat philtrum (lip–philtrum score of 4–5 agreed by three observers) was observed in 7/10 patients with FASD, compared with 16/41 of the non-FASD patients, giving an OR of 3.43. The combination of a thin upper lip and short palpebral fissures was also common in this group. One might expect this as the characteristic facial features form a major part of the diagnostic criteria. In ARBD and ARND, facial features need not be present for diagnosis.6 ,7 The hockey stick creases and railroad-track ear, features mentioned frequently in the literature, did not appear to be reliable markers of FAS14 and there is currently a debate about their usefulness in the diagnosis of FAS. In one study, paediatricians who had been trained to recognise hockey stick creases consistently overestimated their presence compared with clinical geneticists.18 Railroad track ear is a term used to encompass several different abnormalities of the horizontal and inferior crura of the helix and antihelix of the ear. Following recent standardisation of terminology for describing morphological abnormalities, this term has now been made obsolete.19
Genetic referral did not impact significantly on the general health of the child as medical and developmental input was usually already in place through a community paediatrician (40%) or a general paediatrician (22.5%). The consultation was of value in providing information to carers, many of whom had not come into contact with FAS before. We hypothesised that improving the carers' comprehension of the condition would facilitate meeting the child's needs. We could not assess the impact of the consultation where a child had been referred specifically to help with placement for adoption as we had no follow-up information. We envisage that the assessment would have been of some value to prospective adoptive parents. Adoptive parents were often interested in the long term implications of FASD and in particular implications for education. Prenatal alcohol exposure can cause a spectrum of central nervous system consequences that can ultimately result in a severely impaired individual20 and is associated with an increased risk of conduct disorder in adolescence.21
Strength of study
This study is a comprehensive review of a significant cohort of patients seen by clinical geneticists with a referral diagnosis of FASD over a 7-year period. It takes into consideration all aspects of the history, clinical examination and investigation related to the referral of a patient for suspected FASD. As we use a standard pro forma for the recording of information from each genetic consultation, we had comprehensive data on each patient and had access to a large number of clinical photographs. Where photographs were missing this was because consent had not been obtained for photography or because the child had been deemed non-dysmorphic. Consensus opinion between a group of consultant and trainee clinical geneticists was used to arrive at a final diagnosis. The study cohort was large enough to permit comparison of the different diagnostic categories. This study draws attention to the benefits of considering microarray analysis in the assessment of FASD.
Study limitations
This study does not provide complete ascertainment of every patient seen in the department with FASD, as some will have had alternative referral diagnoses. One reason for this survey was that we perceived assessment of these children as taking up a significant amount of clinic time and we wanted to ascertain if we were impacting on their management or whether this type of assessment might be better undertaken by a community paediatrician. We have shown that clinical geneticists have an important role in excluding differential diagnoses, identifying familial implications and providing information and support.
This retrospective study was limited by using a review of standard medical records. We improved the objectivity of our case-note review by defining specific terms, for example, impact on medical management and by reaching consensus opinions on presence or absence of specific features. It was not possible to estimate with any accuracy the diagnostic impact of microarray testing since it is a new technique and was carried out only in a minority of patients, though this has been highlighted as an area for future evaluation. It is likely that microarray analysis will soon be available to the paediatric team as a preferred investigation and children with submicroscopic chromosomal anomalies will be identifiable before referral to the genetic service. Though we have shown benefits of a genetic assessment, access to genetic services is not always readily available and may be not be able to be carried out within the time-frame required for decisions to be made about care placements. Though improved education about FASD and the ability to exclude other conditions with more certainty may lead to increased and more accurate diagnosis, the question of who is best placed to manage affected individuals remains. This is particularly so in the case of adults with FASD for whom few services exist. Prevention of the condition must remain the major goal.
Acknowledgments
The authors would like to acknowledge all the staff from Genetic Medicine, St. Mary's Hospital, Central Manchester University Hospitals NHS Foundation Trust.
References
Footnotes
-
Competing interests None.
-
Funding JCS is supported by the Manchester Biomedical Research Centre.
-
Provenance and peer review Not commissioned; externally peer reviewed.