Article Text

Abstract
Objective To study the epidemiology of diseases that cause progressive intellectual and neurological deterioration (PIND) in UK children.
Design Since May 1997, the authors have performed active surveillance to search for variant Creutzfeldt–Jakob Disease (vCJD) among the many diseases that cause neurological deterioration in children, using the monthly surveillance card sent to all UK consultant paediatricians by the British Paediatric Surveillance Unit. The authors obtain clinical details from reporting paediatricians by questionnaire or site visit, and an Expert Group then independently classifies the cases.
Results After 12 years, 2636 patients less than 16 years old with suspected PIND had been reported, of whom 1114 had a confirmed diagnosis to explain their deterioration: in these children, there were 147 different diseases. These were the six commonest diagnostic groups: leukoencephalopathies (183 cases), neuronal ceroid lipofuscinoses (141 cases), mitochondrial diseases (122 cases), mucopolysaccharidoses (102 cases), gangliosidoses (100 cases) and peroxisomal disorders (69 cases). Relatively large numbers of PIND children were reported from parts of the UK where there are high rates of consanguinity. Only six children with vCJD (four definite, two probable) had been identified.
Conclusions Although this study does not ascertain all UK cases, it provides a novel insight into the epidemiology of the neurodegenerative diseases that cause PIND in children. It is reassuring that in general these children are carefully investigated and that active surveillance has found only six children with vCJD. However, there is concern that more childhood vCJD cases may appear, possibly with a different genotype from those identified so far.
Statistics from Altmetric.com
Introduction
In 1996, Will et al reported 10 cases of a new variant of Creutzfeldt–Jakob Disease (vCJD), with an earlier onset than sporadic CJD and a novel neuropathological phenotype:1 since then, the National Creutzfeldt–Jakob Disease Surveillance Unit (NCJDSU) in Edinburgh has performed surveillance for vCJD, and by January 2010, the total number of definite and probable cases of vCJD in patients of all ages was 170 (166 dead and four alive).2
Since 1997, we have used the active national surveillance mechanism of the British Paediatric Surveillance Unit to perform surveillance for vCJD in children.3 There were three main difficulties in planning this surveillance: (1) there is no diagnostic test for vCJD, (2) the clinical presentation of vCJD in young children might be different from that in adults, and (3) there are many rare diseases that cause neurological deterioration in childhood. We therefore decided to perform surveillance for all children with ‘progressive intellectual and neurological deterioration (PIND),’ because we hoped that such a group would include all children with vCJD.
What is already known about this topic
By January 2010, the total number of definite and probable cases of vCJD was 170; six were less than 16 years old at the onset of symptoms.
The only way to make a definite diagnosis of vCJD is by studying brain tissue.
Although diseases that cause neurological deterioration in children are individually rare, there are many such diseases, and vCJD cases could be hidden among them.
What this study adds
The study provides unique data about the distribution and clinical features of the many disorders that cause PIND in children.
Only a small proportion of the children who died of neurodegenerative disease underwent post-mortem, although before death most children with PIND were carefully investigated.
It is reassuring that no children with vCJD have been identified since 2000, but more cases could occur.
The advantage of studying a relatively broad group of children has been that we have actively and prospectively obtained clinical information about the many diseases that cause PIND in children, and it is the purpose of this paper to report these novel epidemiological findings.
Methods
British Paediatric Surveillance Unit (BPSU)
The BPSU office in the Royal College of Paediatrics and Child Health sends a monthly surveillance card listing the conditions currently under surveillance to all consultant paediatricians in the UK. They are asked to return the card, reporting cases seen in the previous month. Between 2800 and 2900 cards are sent out each month, and over 90% are returned. The BPSU office informs the surveillance groups about reported cases, and they obtain clinical information from the notifying paediatricians. The methodology has been described in detail elsewhere.4 5
PIND Study
Paediatricians are asked to report children with evidence of PIND—see table 1 for the detailed case definition. Information from the local paediatrician is obtained by telephone interview or site visit to extract clinical information from the hospital notes. Data that identify patients are kept secure by the surveillance team. Clinical information is extracted, patients' names are removed, and these data are held on a password-protected computer database.
Case definition—progressive intellectual and neurological deterioration
A PIND Expert Group of paediatric neurologists, a clinical geneticist and a representative from the NCJDSU meets quarterly with the PIND surveillance team to study the anonymised clinical data, review the diagnoses that have already been made by the notifying clinicians and provide feedback about those children who meet the criteria for PIND but have not yet been diagnosed. Any newly presenting case with features suggestive of vCJD would be discussed with the notifying paediatrician, with a recommendation that a referral be made to the NCJDSU which has the expertise to diagnose vCJD and makes contact with the family if consent is obtained from them.
Consent for the PIND study was obtained from the Cambridgeshire 2 Research Ethics Committee (ref: 97/010) and from the Public Health Laboratory Service Ethics Committee. The research was approved by the Cambridge Research and Development Committee and the national Patient Information Advisory Group, now the National Information Governance Board (PIAG/BPSU 2-10(c)/2005).
Results
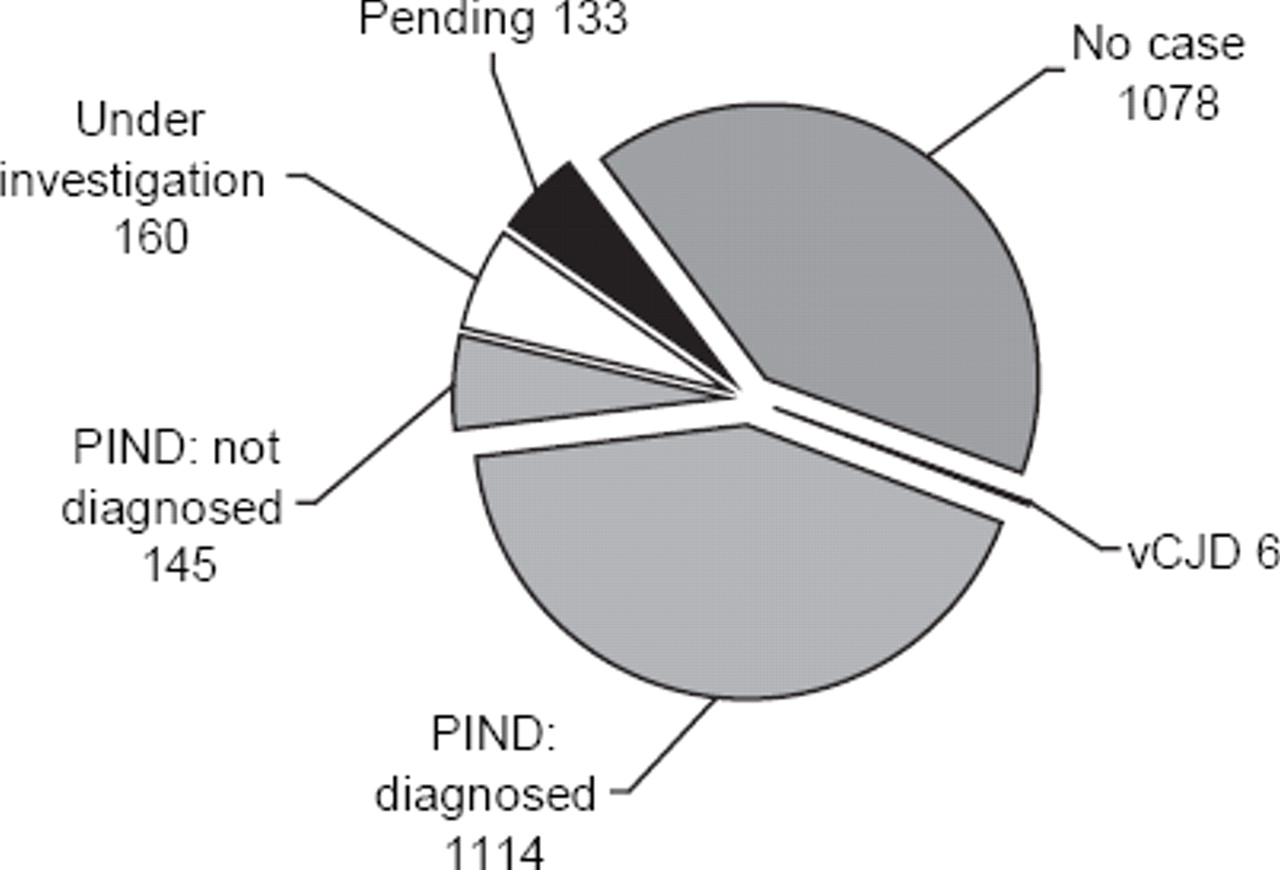
Surveillance for PIND started via the monthly BPSU card in May 1997 and continues. By April 2009, after 12 years of active surveillance, 2636 cases of suspected PIND had been reported (see figure 1). Data had not yet been collected in 133 pending cases, and 1078 were ‘no cases’ (not fulfilling the criteria for PIND, duplicate notifications, reported in error or no traceable clinical information). One hundred and sixty were still under investigation, but so far none of these had the clinical picture of vCJD.

Classification of cases reported to the progressive intellectual and neurological deterioration (PIND) study between April 1997 and May 2009. vCJD, variant Creutzfeldt–Jakob Disease.
Definite or probable cases of vCJD
Only six cases of vCJD (four definite and two probable) had been notified: the youngest was a girl aged 12 years at onset. There were three other girls (aged 13, 14 and 15 years at onset) and two boys aged 15 years at onset. The last child to develop symptoms did so in 2000. All had died, and neuropathology confirmed vCJD in four cases; an autopsy was not performed in the remaining two cases, but they met the criteria for probable vCJD.
Children with a diagnosis other than vCJD
Of the other children who met the PIND case definition, 1114 children had an underlying diagnosis that explained their deterioration. If all the specific diagnostic variants were counted separately, there were 147 known neurodegenerative conditions that we had identified in children with PIND, which illustrates the complexity of classifying such children. The 30 most commonly identified diseases are shown in table 2. Some of these diseases can be placed in diagnostic groups, and the six largest of these groups are shown in figure 2. Each group includes many different disorders, for instance in the neuronal ceroid lipofuscinosis (NCL) group, there were 141 cases with 13 different diagnoses—four types of infantile NCL, four types of late infantile NCL, four types of juvenile NCL and two cases of another NCL variant.
Thirty most commonly reported confirmed diagnoses

{kind=link}
{kind=link}
Progressive intellectual and neurological deterioration cases with definite diagnoses: the six most commonly reported disease groups. L-encephalopathies, leukoencephalopathies; NCLs, neuronal ceroid lipofuscinoses; Mitochondrial, mitochondrial diseases; MPSs, mucopolysaccharidoses; Peroxisomal, peroxisomal disorders.
Children with PIND and no underlying diagnosis
One hundred and forty-five children had deteriorated or died without a diagnosis. This group gives cause for concern because if a ‘new’ variant of vCJD were to arise or if the paediatric presentation differed from the adult presentation, the idiopathic group could include such a phenotype. However, the Expert Group was satisfied that the children in this undiagnosed group had been fully investigated or had a sibling with a similar disease pattern who had undergone extensive investigations. These children were clinically heterogeneous, and none had a clinical presentation suspicious of vCJD; nor was there any evidence of a ‘new’ unrecognised disorder in this group. A more detailed report on these cases has been published.6
Geographical distribution of PIND cases
Particularly large numbers of reported PIND cases live in certain Health Districts. The four Districts with the largest numbers were Bradford (87 cases), Birmingham (67 cases), East London and City (42 cases) and Leeds (38 cases). These compare with a median number of nine cases per Health District when the whole of the UK is considered. In the four Health Districts with the greatest numbers, a large proportion of the PIND children came from consanguineous families of Pakistani or Indian origin (Bradford 71%, Birmingham 52%, East London and City 52% and Leeds 42%). This continues a trend that we have previously reported.7
Discussion
Design of the PIND study
Neurological deterioration in children is caused by a complex mixture of disorders, so the identification of vCJD in children is difficult. If we had asked paediatricians to notify ‘suspected cases of vCJD,’ and none had been reported, this might have been because cases had not been recognised by paediatricians. The strategy is therefore to identify all UK children with evidence of ‘PIND’ and then present the clinical information to an Expert Group of paediatric neurologists who classify the cases. In addition to being a method of searching for vCJD, this is a unique national survey of the causes of neurological deterioration in childhood.
The advantage of using the BPSU system is that it is prospective, and it is strongly supported by paediatricians, with excellent overall response rates. The disadvantage is that there may be under-reporting because of forgetfulness, reporting fatigue or when cases are not seen by a paediatrician. However, in the PIND study, under-reporting is minimised because children with neurological deterioration are often seen by both local and tertiary paediatricians, increasing the chance of notification. While 34% of PIND cases were initially reported by paediatric neurologists, 32% were reported by general paediatricians, 22% by community paediatricians and 12% by other specialist paediatricians.
Epidemiology of neurodegenerative diseases in childhood—strengths and weaknesses of the study
Knowledge about these disorders has previously been based on case series published by individual specialist centres. The PIND data provide a picture of a less selected group of children because they are notified by general and community paediatricians and not just by tertiary specialists.
We are careful to stress that these data do not provide a means of determining accurate incidence or prevalence rates for individual diseases. This is because there is under-reporting of cases for various reasons—busy paediatricians may not be sufficiently motivated to report cases, and they may forget about relevant cases seen in the previous month. They often do not realise that we want to hear about children who fit the case criteria for PIND, even if a specific neurological diagnosis has already been made—we want the PIND Expert Group to have the opportunity to review those diagnoses so we can be as sure as possible that vCJD is not being missed.
Some diseases do not always cause progressive deterioration, and therefore not all cases will be reported. For instance, children with Rett syndrome fit the PIND criteria when their development slows, and they lose skills towards the end of the first year of life. Later, they plateau and are less likely to fit the PIND case definition, so we know that our study has identified only a proportion of children with Rett syndrome.
We include children as cases if they have been diagnosed as having a disease that may cause PIND in the future. For instance, children diagnosed as having adrenoleukodystrophy are included in our numbers, even if they have not yet deteriorated neurologically: indeed, some may never develop neurological problems. Similarly many children with ataxia telangiectasia develop abnormal neurological signs but show no intellectual deterioration. However, they may develop learning problems later, and we have therefore included them (see table 2).
Our study has yielded valuable information about the clinical presentation and mode of diagnosis of the many diseases that cause progressive intellectual and neurological deterioration. This paper summarises the main points about this large group of complex disorders but does not go into great detail about each disease. We have already published our case series of suspected Alpers disease,8 a review of the mitochondrial cases is awaiting publication, and more will follow.
Case for continuing PIND surveillance in children
By January 2010, 170 cases of definite and probable vCJD had been identified in UK patients of all ages.2 The youngest UK child diagnosed as having vCJD developed symptoms in 2000 at the age of 12 years. However, it remains of great public health importance to continue PIND surveillance for the following reasons.
Because of the measures taken to remove potentially infected beef products from the market, it is likely that no child born after 1996 has been exposed to this source of infection, but this means that cases could appear in those aged less than 16 years until 2012.
Until recently, all cases of vCJD were methionine homozygous (MM) at codon 129 of the prion protein gene (37% of the UK population have this genotype). However, recently a possible adult case has been reported with a methionine/valine (MV) genotype (51% of the UK population have this genotype) (R G Will, NCJDSU, personal communication). There is the possibility that more MV cases will appear, possible after a longer incubation period and with a different clinical phenotype in children from that in adults.
There is a risk that children could be infected via blood transfusion, as there are four instances of probable transfusion transmitted infection in adults.9 Three of these developed vCJD; in the other case, there was evidence of infection in the spleen but not in the brain:10 this preclinical case was heterozygous (MV) at codon 129 of the prion protein gene.
There is the potential for vCJD infection linked to treatment of the paediatric population with plasma products—for example Factor VIII.
There is also a potential risk of vertical transmission of infection from mother to child but so far there is no evidence of this.11
An important and reassuring message is that most children notified to us are being investigated very thoroughly by their secondary and tertiary paediatricians. However, only a very small proportion of those who die undergo autopsy (only four out of 46 who died in our undiagnosed group6). Fortunately, the PIND Expert Group agreed that most of the children who died had undergone the appropriate brain scans and other tests before death. Nevertheless in the absence of a diagnostic test for vCJD the only way to be certain that a child has not died of vCJD is to obtain brain tissue for neuropathological examination. Because such a small proportion of children with neurodegenerative diseases undergo post-mortem examination PIND surveillance remains the only systematic way to look for cases of vCJD that might be hidden among the many disorders that cause neurological deterioration in children.
Acknowledgments
Many thanks to the British Paediatric Surveillance Unit of the Royal College of Paediatrics and Child Health and to R Lynn, the Scientific Coordinator. Past and present members of the PIND Expert Group have been an invaluable resource and have been extremely generous with their time; they include J Aicardi, P Baxter, Y Crow, C De Sousa, S Green, J Livingston, M Pike, R Robinson, R Surtees, J Wilson and S Zuberi. The study could not have been performed without the hundreds of paediatricians who have reported cases and have then completed forms and undergone telephone interviews.
Footnotes
-
Funding This is independent research funded by the Department of Health, UK (grant reference: 121/6443). The views expressed in the publication are those of the authors and not necessarily those of the Department of Health.
-
Competing interests None.
-
Ethics approval Ethics approval was provided by the Cambridgeshire 2 Research Ethics Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Patient consent The Patient Information Advisory Group, now the National Information Governance Board, gave the study approval to obtain clinical information about cases without prior approval from carers or patients.