Article Text

Abstract
Background: The effects of maternal phenylalanine on the fetus include facial dysmorphism, microcephaly, intrauterine growth retardation, developmental delay, and congenital heart disease.
Aims: To evaluate the impact of phenylalanine restricted diet in pregnant women with phenylketonuria (PKU) on their offspring.
Methods: Data on virtually all pregnancies of women with PKU in the United Kingdom between 1978 and 1997 were reported to the United Kingdom PKU Registry. The effect of the use and timing in relation to pregnancy of a phenylalanine restricted diet on birth weight, birth head circumference, the presence or absence of congenital heart disease (CHD), 4 year developmental quotient, and 8 year intelligence quotient were examined.
Results: A total of 228 pregnancies resulted in live births (seven twin pregnancies were excluded). In 110 (50%), diet started before conception. For this group mean (SD) birth weight was 3160 (612) g, birth head circumference 33.6 (1.9) cm, 4 year DQ 108.9 (13.2), 8 year IQ 103.4 (15.6), and incidence of CHD was 2.4%. In comparison, for those born where treatment was started during pregnancy (n = 91), birth weight was 2818 (711) g, birth head circumference 32.7 (2.0) cm, 4 year DQ 96.8 (15.0), 8 year IQ 86.5 (13.0), and incidence of CHD was 17%. Month-by-month regression analyses suggested that metabolic control by 12–16 weeks gestation had most influence on outcome.
Conclusions: Many features of the maternal PKU syndrome are preventable by starting a phenylalanine restricted diet. Women with PKU and their carers must be aware of the risks and should start the diet before conception, or as soon after as possible.
- CHD, coronary heart disease
- DQ, development quotient
- IQ, intelligence quotient
- PKU, phenylketonuria
- maternal phenylketonuria
- phenylalanine
- pregnancy
- outcome
Statistics from Altmetric.com
Newborn screening for phenylketonuria (PKU) has been one of the great success stories of preventative medicine in the 20th century.1 This inherited defect of the phenylalanine hydroxylase gene, untreated, results in severe learning disability and behavioural problems associated with medical problems such as eczema and epilepsy. With dietary intervention to restrict phenylalanine intake, it has been converted to one with essentially normal development and social integration. Increasing numbers of people with PKU are reaching adulthood, able to work and form relationships. However, over the past 40 years it has become apparent that the offspring born to mothers with PKU are at risk of damage from the teratogenic effects of phenylalanine.2 High maternal phenylalanine concentrations are associated with a distinct syndrome: facial dysmorphism, microcephaly, developmental delay and learning difficulties, and congenital heart disease.3 A Medical Research Council Working Party recommended that women with PKU should commence a phenylalanine restricted diet preconceptually to protect against these effects.4 Since the mid-1980s in North America a prospective International Collaborative Study5 has collected data on pregnancies of women with PKU, both treated and untreated. Here we report the experiences of maternal PKU within the United Kingdom from data collected within the UK Phenylketonuria Registry.
METHODS
The Phenylketonuria Register supported by the Medical Research Council has collected information on virtually all known females with PKU born in the United Kingdom since 1964. A few were notified because problems with their offspring led to the diagnosis. The original aims of the study were to alert clinicians caring for these women about the risks of maternal PKU; to provide an overall picture of the situation in the UK regarding maternal PKU; and to collect data to compare the outcome of pregnancies in women receiving dietary intervention at different stages of their pregnancies. The main outcome measures were birth weight, birth head circumference, development (DQ)/intelligence quotients (IQ) at 4 years6 and 8 years (WISC-R7), and the presence or absence of congenital heart disease. Interim reports have been published previously.8,9 Approval for analysis of the data was obtained from the Research Ethics Committee of the Institute of Child Health, London.
Statistical methods
Analysis of variance was used to compare the outcome measures, birth weight, head circumference, DQ, and IQ at ages 4 and 8, between the three diet groups: no diet, preconception diet, and started diet during pregnancy. Relations between these outcome measures were investigated using Pearson’s correlation coefficient.
For the phenylalanine data, only measurements obtained on visits made between 100 days preconception and 300 days of gestation were considered in the analyses. Blood phenylalanine concentrations were measured and scored as follows: 1 = <120 μmol/l; 2 = 120–299 μmol/l; 3 = 300–599 μmol/l; 4 = 600–899 μmol/l; 5 = 900–1199 μmol/l; 6 = 1200–1499 μmol/l; 7 = 1500–1799 μmol/l; 8 = 1800–2099 μmol/l; 9 = 2100–2399 μmol/l; 10 = >2400 μmol/l. Although the data were recorded on an ordinal scale 1–10, this represents a linear transformation of the original units of the phenylalanine measurements. Thus the data were used as a continuous measurement.
For each pregnancy, the average phenylalanine concentration for each of the nine months during pregnancy and three months before conception were calculated. Only pregnancies where data were available for all nine months of pregnancy were included. Regression analysis was then used to investigate the strength of the relation between each of the average monthly phenylalanine levels with both birth weight and head circumference. An adjustment was made for known predictors, sex, and gestation (quadratic effect). Since the data were not independent, as some pregnancies were for the same women, robust standard errors10 were used when testing the significance of the predictors. The Normality assumption of both analysis of variance and regression analysis was checked using the Shapiro-Francia W dash test.11 Bartlett’s test was used to check for equality of variances between groups for ANOVA. Analyses were carried out using the statistical software package Stata.12
RESULTS
Between 1978 and 1997, 155 women with PKU had 228 pregnancies resulting in live births. Seven pregnancies resulted in twins and were excluded from the analyses. Data were available for birth weight in 205 pregnancies, for head circumference in 197, for cardiac status in 172, for DQ at 4 years in 50, and for IQ at 8 years in only 32 pregnancies. Complete data were available for relatively few pregnancies.
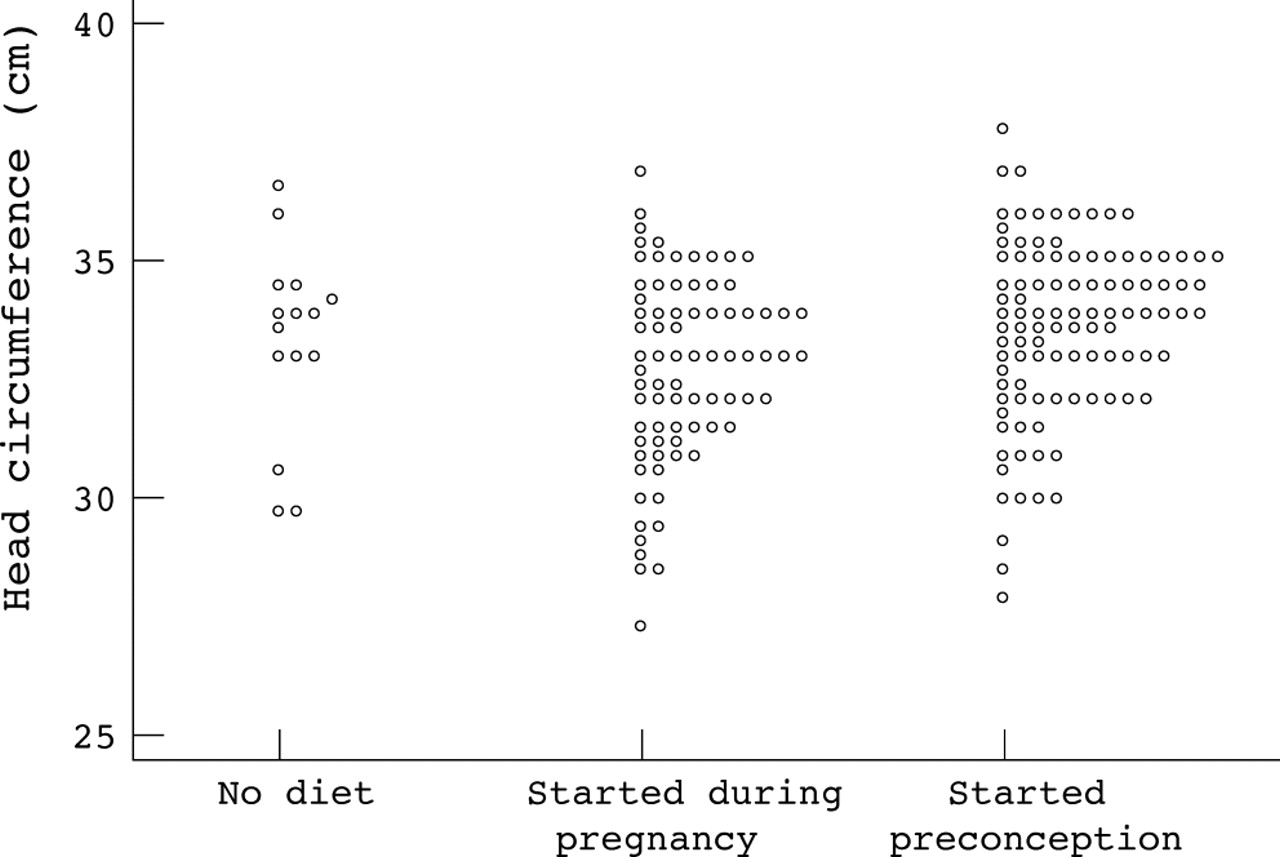
For the primary analyses, the pregnancies were divided up into three diet groups: phenylalanine restricted diet never commenced (n = 18, 8%), diet commenced at some stage during pregnancy (n = 91, 42%), and diet started before conception (n = 110, 50%). For two pregnancies dietary regimen was unknown. It is unclear why diet was never commenced in the 18 women. They might have been too intellectually disabled to manage diet but capable of refusing termination of pregnancy; they may have presented too late in pregnancy; or possibly their clinicians may have been unaware of the need for diet. The outcomes for birth weight and birth head circumference are shown in figs 1⇓ and 2⇓ respectively. Mean (SD) birth weight for those never treated was 2978 (436) g, for those started during pregnancy 2818 (711) g, and for those on preconception diet 3160 (612) g (p = 0.002 from ANOVA). Mean (SD) birth head circumference for those never treated was 33.3 (2.1) cm, for those started during pregnancy 32.7 (2.0) cm, and for those on a preconception diet 33.6 (1.9) cm (p = 0.01 from ANOVA). Babies born to women who started the diet preconception had, on average, a larger birth weight and head circumference, compared with babies born to women starting diet later (p < 0.001 for both). There was no evidence of such differences for babies whose mothers were never treated compared to either of the other groups. Congenital heart disease was present in 12/72 (17%) of infants whose mothers started the diet during pregnancy and in only 2/84 (2%) of those born after the diet was started preconceptually. The difference is 14% (95% CI 5–23%, p = 0.002 from χ2 test. For those infants whose mothers were never on the diet, congenital heart disease was present in 1/15 (7%).

Birth weight of infants born to women with phenylketonuria according to timing of dietary intervention.

Birth head circumference of infants born to women with phenylketonuria according to timing of dietary intervention.
Looking at DQ at 4 and IQ at 8 years, there was good correlation between the two outcome measures (r = +0.88, p < 0.001). Weaker correlations were present for DQ at 4 years and birth weight (r = +0.32, p = 0.03) and birth head circumference (r = +0.43, p = 0.003); and IQ at 8 years and birth weight (r = +0.37, p = 0.05) and birth head circumference (r = +0.49, p < 0.01). The impact of timing of diet and DQ or IQ at 4 and 8 years is shown in figs 3⇓ and 4⇓ respectively. Mean (SD) DQ at 4 years for those never treated was 72.3 (25.1), for those started during the pregnancy 96.8 (15.0), and for those on the preconception diet 108.9 (13.2) (p < 0.001 from ANOVA). Mean (SD) IQ at 8 years for those never treated was 39.5 (0.71), for those started during the pregnancy 86.5 (13.0), and for those on the preconception diet 103.4 (15.6) (p < 0.001 from ANOVA). For women on a preconception diet, their offspring had significantly higher mean DQ or IQ scores at both ages 4 (p = 0.03) and 8 years (p < 0.01) compared with other two diet groups.

McCarthy Developmental Quotients at 4 years in infants born to women with phenylketonuria according to timing of dietary intervention.

WISC-R Intelligence Quotients at 8 years in infants born to women with phenylketonuria according to timing of dietary intervention.
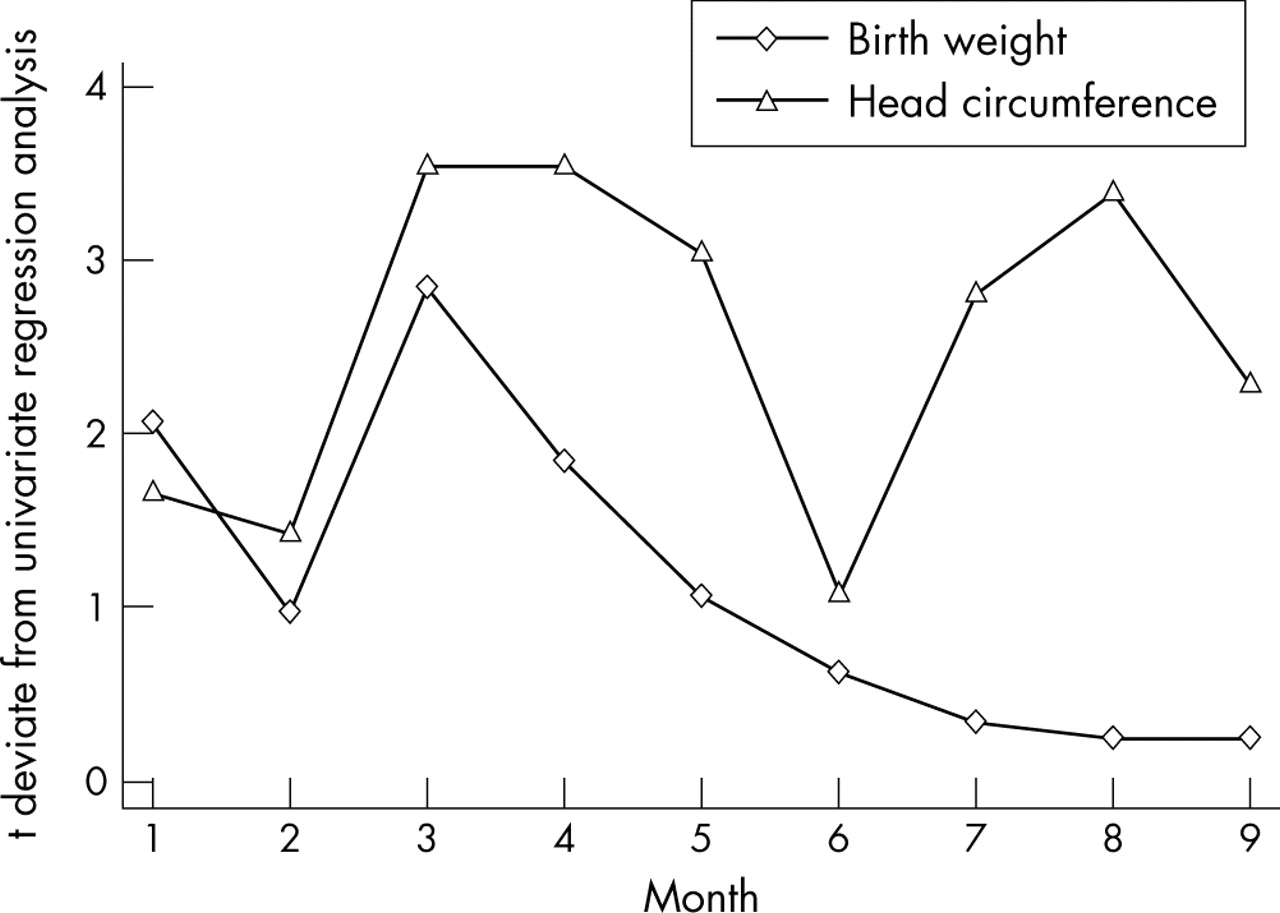
Blood phenylalanine concentrations were measured between 1 and 165 times for each pregnancy (mean 56, median 40). Because of missing data for DQ and IQ, blood phenylalanine has been examined only in relation to birth weight and birth head circumference. Average monthly phenylalanine levels were examined month-by-month using a regression equation corrected for gestation and sex. The t deviate values for these analyses, defined as the ratio of the estimated slope coefficient from regression analysis to its standard error are shown in fig 5⇓. The higher the t deviate, the stronger the influence that blood phenylalanine concentration has on the outcome measure. For all analyses, the direction of the relation between average blood phenylalanine concentration and outcome was negative, suggesting that for higher concentrations, predicted birth weight decreases. Average phenylalanine seemed the most important for birth weight at month 3 (12 weeks gestation) and between months 3 and 4 (12–16 weeks gestation) for birth head circumference.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
t deviate values for birth weight and birth head circumference analysed month-by-month for infants born to women with phenylketonuria.
Five clinics cared for the pregnancies of between 12 and 55 live births. Eighty seven centres looked after less than eight pregnancies during this time. Comparing birth weight and head circumference between the centres depending on their relative experience of maternal PKU, mean (SD) birth weight was lower (2906 (671) g v 3076 (647) g, p = 0.07) and head circumference was smaller (32.8 (2.12) cm v 33.4 (1.81) cm, p = 0.04) in centres with least practical experience of managing these pregnancies, although there was considerable spread of data.
DISCUSSION
Despite a recent consensus statement from the National Institutes of Health in the United States,13 many questions remain about the management of phenylketonuria into adult life. The only situation where it is quite clear that dietary intervention is of benefit, is to protect the unborn fetus of women with PKU.5 The data reported here from a cohort of women with PKU from the United Kingdom support the need for the early introduction of a phenylalanine restricted diet for these pregnancies and for management of these pregnancies in centres with experience. Neonatal birth weight and head circumference are greater, the incidence of congenital heart disease is lower, and developmental and intelligence quotients are higher at 4 and 8 years of age in those offspring whose mothers went on the diet before they were born. Nevertheless, a number of questions about maternal PKU also need to be answered. These include the effects of introducing the diet after conception, what concentrations of blood phenylalanine should be aimed at, whether or not different quality diets affect outcome, the impact of both fetal and maternal genotype, and the effects of the postnatal environment, including parental IQ.
Unfortunately, the data obtained from the PKU Registry can only help with answering the first question. Over 90 centres provided variable information about their patients and pregnancies, as resources to support their collection were limited. The quality of these data makes it impossible to evaluate either precise blood phenylalanine concentrations or differences in dietary therapy during the pregnancies. As a result much of the data for each pregnancy was incomplete. These and similar challenges face many clinical research workers as they consider establishing disease registers today. In addition, the register was started at a time when molecular genetics knowledge was limited, so genotyping of mothers and/or offspring was not performed.
Nevertheless, the data have revealed some interesting findings. Although as a group, the children born to women who started diet prior to conception did better than those starting diet when pregnant or not at all, the overlap in the outcomes shown in figs 1⇑–4⇑, show that this is not always the case. Indeed some of the children from untreated pregnancies did remarkably well, while some from those on the diet preconceptually and throughout the pregnancies did poorly. Close examination of these particular cases may reveal important clues regarding factors protecting against the teratogenic effects of phenylalanine, as well as the detrimental effects of too low phenylalanine concentrations.14 Further work regarding the regulation of placental transport of phenylalanine and other nutritional factors, including the role of hypotyrosinaemia, is necessary.15 However, the group that were never treated was small and perhaps selected themselves out compared to other untreated pregnancies that were terminated as was recommended at the time.4 The marked diurnal fluctuations in blood phenylalanine concentrations in PKU have only relatively recently become apparent.16 The MRC recommendations are for twice weekly phenylalanine measurements before pregnancy and thrice weekly after conception,4 but even these may be a poor indication of what is really happening. In the United States only weekly blood sampling is recommended for logistic reasons. The similar results between the United Kingdom and United States maternal PKU cohorts suggest this may be sufficient.
Within the National Health Service, care for patients is currently free at the point of delivery. This means that the challenge that clinicians face with women with PKU at risk of pregnancy is to keep in contact with them throughout their lives. The women can then be supported in decisions they make about their lives and in particular when to start a preconception phenylalanine restricted diet. This is very different from the diet recommended to adults with PKU as the target ranges currently recommended for phenylalanine are between 100 and 360 μmol/l4,5 rather than up to 700 or 1000 μmol/l.4,17 However, the lack of resources in terms of transition programmes from paediatric to adult departments and services for adults with inborn errors of metabolism mean that women with PKU who become pregnant on an unrestricted diet are still seen as they are not in regular contact with a metabolic clinic. Data from the UK PKU Registry suggest that the outcome of offspring is better in the pregnancies cared for by centres with more experience of these pregnancies. In other countries where health insurance companies sanction and pay for therapies, the limited resources are compounded by financial constraints. Many US insurance companies refuse to pay for the diet until the women become pregnant.18 The question of when to start diet after conception becomes increasingly important in these situations. Recent data from the North American Collaborative Study19 have suggested that outcome can be satisfactory when diet is commenced before or up to 10 weeks after conception. The United Kingdom data we report tend to support this, with starting diet before 12–16 weeks gestation strongly influencing outcome. One might expect this to protect brain development, but it leaves embryogenesis of the heart unprotected as this is complete by 10 weeks.20 The influence of blood phenylalanine on head circumference also seems to be important in the third trimester (see fig 5⇑) which perhaps is not surprising given the known impact of hyperphenylalaninaemia in infancy. It suggests that it is worthwhile lowering maintaining blood phenylalanine at this stage in pregnancy to protect fetal brain development.
Examining the rates of preconception diet commencement provide valuable information about healthcare provision. In Lenke and Levy’s seminal survey of maternal PKU in 1980,3 only 3 of 524 (0.6%) pregnancies had diet started prior to conception; in the prospective North American Collaborative Study this increased to 26%;5 in the UK PKU Registry this was 146 of 479 (30.5%) pregnancies. A single centre dedicated to the care of adults with inborn errors of metabolism within the United Kingdom was able to increase this figure to 46 of 59 (78%).21
In summary, analysis of data regarding the pregnancies and offspring of women with PKU within the United Kingdom over a 20 year period has confirmed the impact that phenylalanine has on fetal development, and that it can be ameliorated by the introduction of a phenylalanine restricted diet at an early stage. Treatment significantly reduces the incidence of congenital heart disease, increases both brain and somatic growth, and improves intellectual outcome at 4 and 8 years of age. In addition, the data suggest that starting dietary therapy before 12–16 weeks gestation protects the fetus during the period of maximum teratogenicity of phenylalanine. The maternal PKU syndrome is preventable, but still occurs because of the lack of appropriate resources to care for at-risk women with PKU. Improved programmes for the transition of females with PKU from paediatric to adult services, more staff and facilities dedicated to the care of adults with inherited metabolic disease, and more secure funding processes are all necessary. How often blood phenylalanine should be measured and the precise targets for blood phenylalanine, as well as other nutrients during pregnancy, are not entirely clear, nor are the reasons why some offspring are spared the harmful effects of phenylalanine. The impact of the postnatal environment in which these infants find themselves also requires further evaluation. Improved understanding of the pathogenesis of maternal PKU is still needed to optimise care for these mothers.
Acknowledgments
We would like to thank Dr Isabel Smith who ran the registry during its existence and all the centres that contributed their data to the registry, which was supported by grants from the Medical Research Council and the Department of Health. The UK Newborn Screening Programme Centre latterly provided direction. We would like to thank the National Society for Phenylketonuria for providing support for the analysis of these data.
REFERENCES
Footnotes
Competing interests: none declared