Article Text

Abstract
A 19 day old infant was successfully resuscitated from ventricular fibrillation. The 12 lead ECG was normal, with a normal QT interval, and remains so over three years follow up. DNA analysis revealed a missense mutation (R1193Q) in the SCN5A gene, previously linked with familial sudden unexpected nocturnal death syndrome, also known as Brugada syndrome.
- ICD, implantable cardioverter defibrillator
- LQTS, long QT syndrome
- SIDS, sudden infant death syndrome
- SUNDS, sudden unexpected nocturnal death syndrome
- VF, ventricular fibrillation
- Brugada syndrome
- cardiac channelopathy
- long QT syndrome
- SIDS
- SUNDS
Statistics from Altmetric.com
- ICD, implantable cardioverter defibrillator
- LQTS, long QT syndrome
- SIDS, sudden infant death syndrome
- SUNDS, sudden unexpected nocturnal death syndrome
- VF, ventricular fibrillation
Data supporting a link between sudden infant death syndrome (SIDS) and sudden cardiac death are scarce. There have been only two near-miss sudden infant deaths reported where torsade-de-pointes or ventricular fibrillation (VF) has been documented and linked to causative genetic changes.1,2 Both infants had long QT syndrome (LQTS) type 3, with mutations in SCN5A, a gene that encodes for the cardiac sodium channel (INa) involved in the cardiac action potential.
We report a 19 day old infant presenting with ventricular fibrillation and a missense mutation within SCN5A that has been reported in a family with sudden unexpected nocturnal death syndrome (SUNDS), which is synonymous with Brugada syndrome.3
CASE REPORT
A 19 day old male infant who had been apparently well, gave a sharp cry in his mother’s arms; his skin looked mottled and pale. Shortly before arrival at the nearby hospital the infant became apnoeic and pulseless, and the mother commenced mouth-to-mouth resuscitation and chest compressions.
On arrival an ECG revealed coarse VF. DC cardioversion was successful on the tenth occasion, using a 30J shock with the paddles in a front-back position. He was extubated 20 hours later and made a full recovery. The birth weight was 4170 g, and the postnatal course was routine. The parents, who had no other children, are non-consanguineous. The mother is of Samoan and the Father of Indian Fijian origin; neither have a history of syncope. Two siblings of the infant’s mother had died unexpectedly; one was an intellectually retarded man who drowned in a swimming pool at the age of 20 years. The other died of an unspecified heart problem, aged 8 months. A further four siblings are well. There was no family history of SIDS. There was no history of drug ingestion by the infant, or his mother.
There were no dysmorphic features. At 24 hours, the heart rate corrected QT interval (QTc; QT/sq root R-R interval) was marginally prolonged (0.48 s, see fig 1). Echocardiogram and cardiac magnetic resonance imaging were normal. Metabolic screening was negative. A cardiac electrophysiological study one month later failed to induce any arrhythmia. Twelve-lead ECGs from the mother and grandmother had a borderline QTc of 0.46 seconds, just below the upper limit of normal for adult females (0.47), and were otherwise normal. The father and maternal grandfather had a normal ECG and QTc. The infant was started on atenolol at a dose of 4 mg/kg/day. His parents were provided with an automated external defibrillator (AED) and an apnoea alarm. Follow up QT intervals have remained within the normal range (fig 1). At the age of 44 months, he is developing normally, he has had no further events, and the QTc is 0.38 sec.

Extracts from the surface electrocardiogram. Left: The day after the cardiac arrest, the T-wave has a late onset and is broad, the QT is measured at a minimum of 0.32 seconds, R-R interval 0.44 seconds, giving a QTc of 0.48 seconds. On some beats the end of the T-wave is less clear, with a long gentle slope into the p-wave such that the true QTc may be a little longer. Right: At 3 months of age the 12 lead ECG is normal. The T-wave is not suspicious for long QT syndrome, and the QT is approximately 0.28 seconds, R-R interval 0.49 seconds, giving a QTc of 0.40 seconds. No features suggest Brugada syndrome.
MOLECULAR GENETIC SCREENING
A LQTS genetic screen4 was performed on the patient and parental DNA, including all five major disease genes KCNQ1, HERG, SCN5A, KCNE1, and KCNE2. All LQTS amplimers were screened using Transgenomics denaturing high performance liquid chromatography (dHPLC) 2100 WAVE DNA Fragment Analysis System and DNASep column (Transgenomic, Santa Clara, USA). Abnormal dHPLC profiles were sequenced by ABI 3100 technology (Foster City, USA).
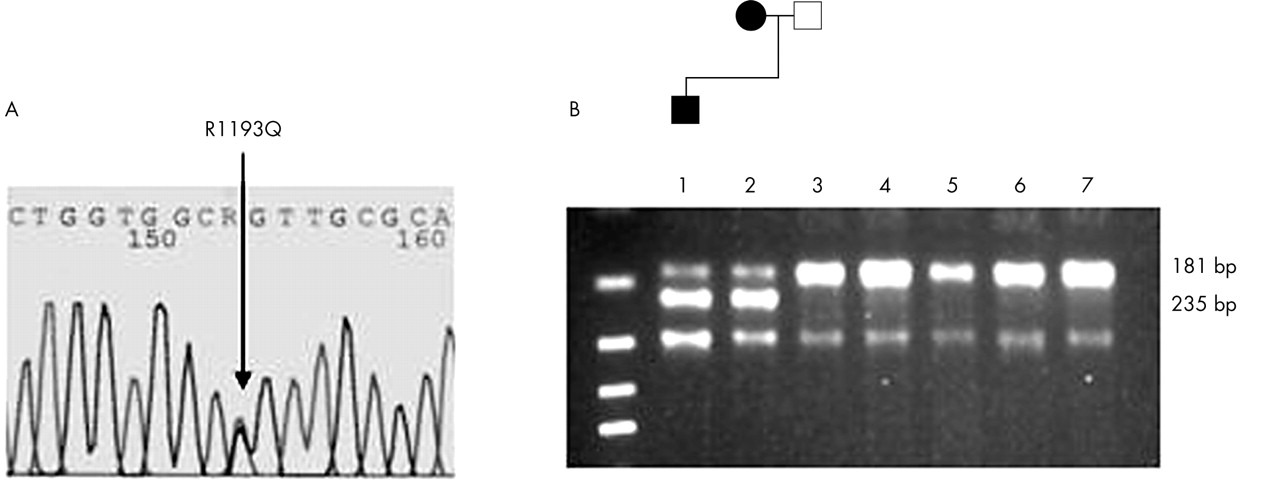
In addition to common polymorphisms, a heterozygous mutation (g3578a) was discovered in position 3578 of the SCN5A coding sequence in the infant and mother (fig 2). The SCN5A g3578a genotype results in a missense substitution of arginine to glutamine at position 1193 of the SCN5A amino acid sequence (R1193Q) which has been detected and functionally assessed in a study detailed by Vatta and colleagues.3 This mutation was subsequently excluded from 180 normal control chromosomes by restriction fragment length polymorphism (RFLP) analysis.
{kind=link}
{kind=link}
Sequence analysis of SCN5A ex20 using patient DNA. (A) The sequence reveals heterozygosity for guanine to adenine (G→A) nucleotide change (arrow) at the second position of codon 1193, which predicts a substitution of arginine by glutamine (R1193Q). (B) AciI restriction enzyme analysis of SCN5A exon 20 in the family members (lanes 1–3) and controls (lanes 4–7). The loss of an AciI site creates fragment sizes of 235bp and 181bp that are visible in the affected child and the maternal side of the family; whereas no 235bp fragments were observed in the paternal side of the family or in 90 control subjects.
DISCUSSION
This is only the third case of sudden infant death where torsade-de-pointes or VF has been documented and linked to a causative molecular pathology. It is the second case where the infant has survived, and the first with a normal resting ECG and a molecular diagnosis of sudden unexpected nocturnal death syndrome (SUNDS). The screening of LQTS genes in 93 SIDS victims revealed pathological SCN5A mutations linked to LQT type 3 in two cases (and one each of LQT types one and two).5 A further SIDS case with ECG indicated LQTS diagnosis prior to death,1 was associated with a SCN5A mutation, while another case was gene positive for LQT type 1.6
Mutations in different positions within SCN5A have also been associated with Brugada syndrome, SUNDS, and idiopathic ventricular fibrillation.3 These conditions are typically associated with sudden unexpected death, mostly in Asian males during sleep, due to ventricular fibrillation or rapid ventricular tachycardia. Recent evidence suggests that SUNDS and Brugada syndrome are in fact the same condition,3 due to reduced function of the cardiac sodium channel INa, an essential determinant in the early phase of the cardiac action potential. Neither have been linked to SIDS previously. Some Brugada cases have a typical ECG appearance of a right bundle branch block pattern in the anterior precordial leads, with early ST segment elevation. The resting ECG can, however, be entirely normal, as in the present case.
Management is difficult since no medications reduce the risk of sudden death. Implantable cardioverter defibrillators (ICDs) are used in adults with Brugada syndrome in survivors of cardiac arrest. We plan ICD placement at age 5 in this child.
Brugada syndrome/SUNDS, with a normal ECG, can cause SIDS. The case highlights the increasing importance of DNA preservation and cardiac channelopathy gene screening in such cases.
Acknowledgments
We are grateful to Dr Russell Snell for the use of genomic equipment based in Vialactia Biosciences. This work was supported by the Child Health Foundation of New Zealand, the Lion Foundation, and the University of Auckland Vice-Chancellor Fund.
Footnotes
-
Competing interests: none declared