Article Text

Abstract
Cerebral oedema is the most common cause of mortality and morbidity during the first day of conventional treatment for diabetic ketoacidosis in paediatric patients. It is possible that therapy contributes to its development. Risk factors that predispose to cerebral oedema should lead to an expansion of the intracellular and/or the extracellular fluid compartment(s) of the brain because water normally accounts for close to 80% of brain weight. With respect to the intracellular fluid compartment, the driving force to cause cell swelling is a gain of effective osmoles in brain cells and/or a significant decline in the effective osmolality of the extracellular fluid compartment. Factors leading to an expansion of the intracerebral extracellular fluid volume can be predicted from Starling forces acting at the blood-brain barrier. Some of these risk factors have an early impact, while others have their major effects later during therapy for diabetic ketoacidosis. Based on a theoretical analysis, suggestions to modify current therapy for diabetic ketoacidosis in children are provided.
- blood-brain barrier
- brain cell swelling
- osmolality
- hyperglycaemia
- insulin
- isotonic saline
- BBB, blood-brain barrier
- CT, computed tomography
- DKA, diabetic ketoacidosis
- ECF, extracellular fluid
- ICF, intracellular fluid
- ICP, intracranial pressure
- NHE, Na+
- H+ exchanger
Statistics from Altmetric.com
- BBB, blood-brain barrier
- CT, computed tomography
- DKA, diabetic ketoacidosis
- ECF, extracellular fluid
- ICF, intracellular fluid
- ICP, intracranial pressure
- NHE, Na+
- H+ exchanger
Cerebral oedema is a potentially devastating complication that occurs in the first day of therapy for diabetic ketoacidosis (DKA).1,2 It is commonly seen 5–15 hours after therapy begins, and there is often little warning that it might develop. Cerebral oedema should be suspected when there is an unexpected deterioration in neurological status or the persistence of a comatose state without an obvious cause. Therefore children with DKA should be admitted to a unit where they can be observed closely for a change in neurological status that includes the development of a headache or vomiting, a diminished ability to respond to questions, or worsening coma.
Cerebral oedema is primarily a clinical diagnosis. The presumptive diagnosis of cerebral oedema is more secure if there is a rapid improvement in neurological status in response to intravenous administration of 3% NaCl or hypertonic mannitol. Making and acting on a clinical diagnosis should take precedence over performing a computed tomography (CT) scan because the latter might delay the implementation of emergency therapy. In fact, confirmatory changes of cerebral oedema on CT scan studies frequently do not keep pace with the clinical course, and they might not be sensitive enough to detect cerebral oedema. Moreover, care might suffer when the patient is transported to an area of the hospital where continuous monitoring may be less than ideal.
Risk factors for cerebral oedema were evaluated recently in two large, carefully conducted, retrospective reviews,1,2 where 61 and 34 patients respectively with cerebral oedema were identified. Cerebral oedema occurred in slightly less than 1% of children treated for DKA—mortality rate was 21 and 24%, respectively. The risk of cerebral oedema appeared to be greater in younger and newly diagnosed diabetics. In the study of Glaser and colleagues,2 a higher plasma urea concentration, a lower arterial pCO2 (calculated from measured venous values with assumptions that are open to question when the arterial value was lacking), and therapy with sodium bicarbonate (NaHCO3) were statistically associated with cerebral oedema. Of interest, their data as well as those of others1,3,4 suggested that a smaller increase in plasma sodium (Na+) concentration (PNa) during therapy may be associated with cerebral oedema.
The lack of a design for ideal treatment of paediatric patients presenting with DKA that minimises the risk of developing cerebral oedema was highlighted in two recent editorials.5,6 In both, the authors implied that current fluid and electrolyte management might contribute to the development of cerebral oedema. Part of the reason for this is that clinicians do not have good data to predict how large the deficits of Na+, K+, and water are likely to be in this population because they can be quite variable in individual patients. These facts make it almost impossible to select an ideal intravenous therapy for these patients. Nevertheless, in this as in other fluid and electrolyte disorders, one need not replace the entire deficit in a precise fashion or at “just the right rate” to minimise risks for the patient. Rather, one must not create a problem by having an inadequate or an excessively large/rapid infusion rate at a time when the patient is most vulnerable. Hence our aim is to provide a theoretical background that might help identify some of these risks, how they might interact, and of greatest importance, emphasise the importance of timing of each therapeutic modality.
For cerebral oedema to become life threatening, intracranial pressure (ICP) must rise appreciably. Five potentially important risk factors that may contribute to the development of cerebral oedema will be emphasised because they may help to explain an increase in the intracerebral intracellular fluid (ICF) and/or extracellular fluid (ECF) volumes (figs 1 and 2). It is obvious that if one of these intracranial compartment volumes were to expand and if this were not accompanied by an equivalent decrease in the volume of the other, there would be a rise in ICP because the brain is enclosed in the skull. Each risk factor must be considered with respect to its magnitude, timing during therapy, and the presence of other risk factors, to understand why a clinically devastating rise in ICP occurred.
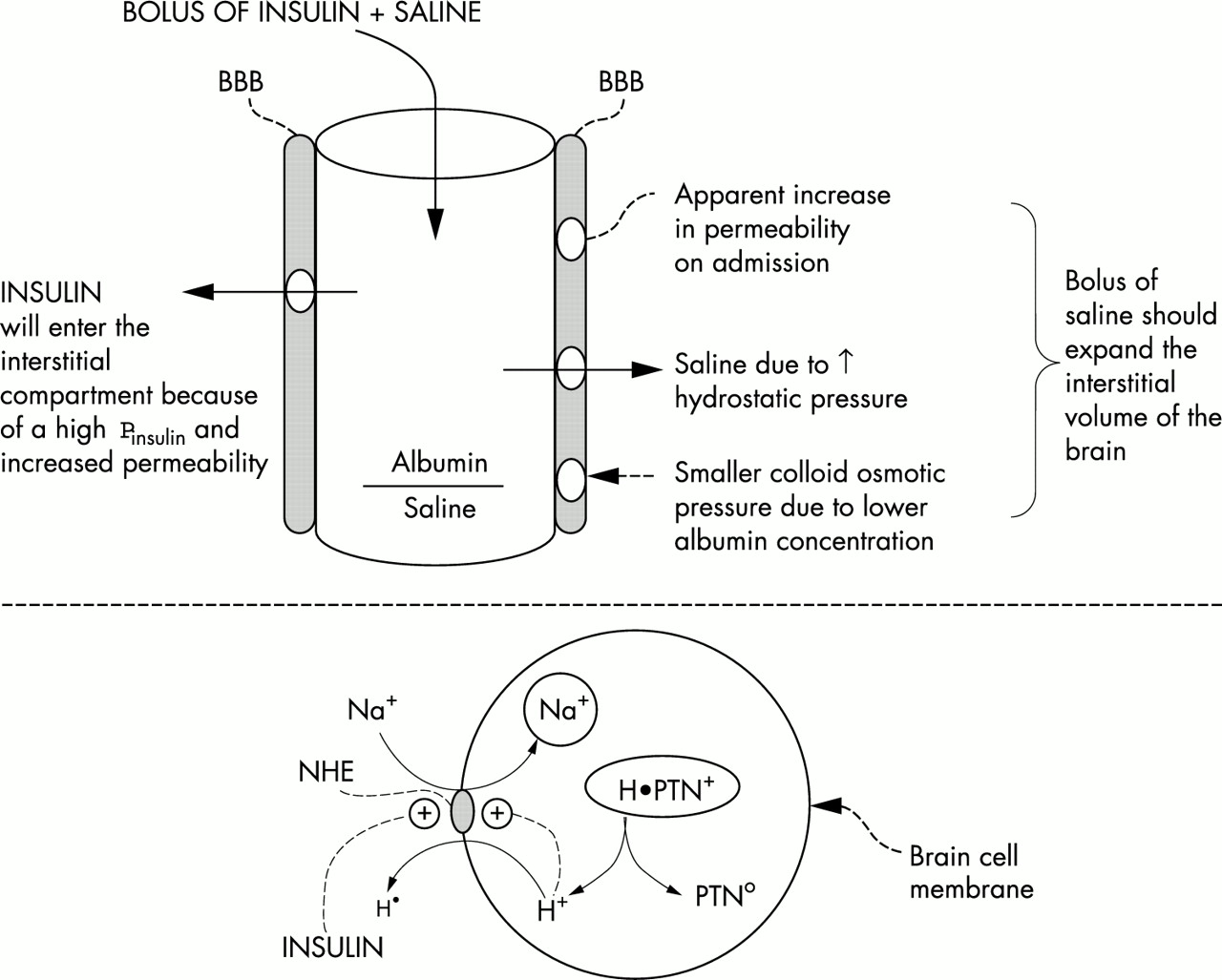
Early risk factors for the development of cerebral oedema. The top portion depicts the BBB that may be less restrictive early in therapy for DKA. Hence a bolus of saline could expand the intracranial interstitial volume. A bolus of insulin could expand the intracerebral ICF volume by converting the inactive form of NHE to its active form (bottom portion)—this causes Na+ to enter and H+ to exit from cells. One ultimate source of H+ in the ICF is from macromolecules (proteins designated H•PTN+). The net result is the electroneutral and stoichiometric exchange of cations (a gain of monovalent Na+ and the change in protein charge form a cationic to a less cationic form, depicted in the ICF ovals).

{kind=link}
{kind=link}
Late risk factors for the development of cerebral oedema. The risk associated with an infusion of too large a volume of saline (left portion) is expansion of the interstitial volume of the brain. If this occurs, the patient may develop an increased ICP even if there is a less severe degree of brain cell swelling. As shown in the right portion, a rise in PNa is needed to prevent a fall in the effective Posm when there is a fall in PGlu. The PNa must be >140 mmol/l if the PNa on admission is close to 140 mmol/l.
There are two major factors that could expand the ICF volume: a rise in the number of solute molecules restricted inside cells (effective osmoles) and a fall in the effective Posm (equation 1) because water moves rapidly across cell membranes to achieve effective osmotic equilibrium. Prior to treatment of DKA, these effective ICF osmoles are primarily potassium (K+), low molecular weight ICF anions, and glucose plus its metabolites. The ECF volume is determined primarily by the number of effective osmoles in this compartment (PNa and the plasma glucose concentration (Pglu)).7 The interstitial compartment of the ECF can rise if there is a higher capillary hydrostatic pressure, a lower plasma colloid osmotic pressure, or capillaries that no longer effectively restrict the movement of albumin. Hence one can anticipate what the major factors leading to cerebral oedema will be.

When assessing changes in the ECF volume, we shall express the rate of administration of intravenous saline as mmol Na+/kg body weight to facilitate comparisons in patients with different body sizes. For reference, if a 20 kg person has close to 4 litres of ECF (20% of body weight) and a PNa of 140 mmol/l, there would be 560 mmol of Na+ per 20 kg in his ECF compartment (close to 30 mmol Na+/kg body weight). Thus a positive balance of 3 mmol Na+/kg expands the ECF volume by 10% if there is little change in the effective Posm. When comparisons are made, 3 mmol Na+/kg is equivalent to 20 ml isotonic saline/kg body weight.
RISK FACTORS FOR CEREBRAL OEDEMA PRIOR TO THERAPY OF DKA
The fact that cerebral oedema is present in close to 5% of patients prior to therapy for DKA1,2 strongly suggests that there are risk factors which might be present before therapy begins. We speculate that one of these factors is that the blood-brain barrier (BBB) might be less restrictive on admission in patients with DKA, because CT scans in several studies have shown a diminished diameter of the ventricular system prior to therapy for DKA.8–10 If this represents brain swelling, it was evident despite a contracted ECF volume (should shrink the brain ECF volume) and a higher effective Posm (should diminish the ICF volume unless there was a large increase in ICF solutes in brain cells).
A fortuitous recent observation could provide insights into why cerebral oedema might develop in a specific patient prior to therapy for DKA.11 We reported data from a 17 year old female who had multiple admissions for DKA because she was not compliant with recommended therapy. On the last admission, she presented with an extremely high PGlu (70 mmol/l (1260 mg/dl)), but she did not have obvious ketoacidosis. She excreted 1 litre of urine in the first 100 minutes in the emergency department. The major basis for her extremely large polyuria was a glucose induced osmotic diuresis that was likely caused by the intake of sweetened fruit juice. Her PGlu at the start and end of this first 100 minute interval remained at 70 mmol/l and her PNa did not change appreciably—hence her effective Posm was virtually constant. When she no longer had access to sweetened fruit juice in the emergency department, she drank tap water. In the next 100 minute period, there was a sudden fall in her effective Posm because her PGlu fell by 30 mmol/l while there was only a minor change in her PNa. This fall in effective Posm could represent a clinically occult basis for the development of cerebral oedema.11 To suspect its presence, a more detailed history of the type of fluid intake is needed in a patient with an extremely large urine output. By finding a change in the ingested fluid from sugar containing fruit juice to tap water, one might be alerted to the possibility that there was an occult fall in the effective Posm (equation 1). Without this detailed history or without having the first two sets of blood values, the physician in the emergency department would not be aware of this potential risk factor for the development of cerebral oedema.
RISK FACTORS FOR CEREBRAL OEDEMA EARLY IN THE THERAPY OF DKA
Infusion of saline
With the background of a less restrictive BBB discussed above, dangers associated with a large intravenous bolus of saline can be anticipated by analysing Starling forces across capillary membranes (fig 1, top portion). An intravenous bolus of saline distributes initially in the plasma volume and reaches the brain; there is little mixing with the large ECF compartment. There are two major reasons for this saline bolus to cause the exit of fluid across the BBB into the interstitial fluid compartment of the brain. First, capillary hydrostatic pressure might be higher because of an expanded plasma volume. Second, the colloid osmotic pressure might fall because of lowering of the plasma albumin concentration by dilution. The colloid osmotic pressure is also influenced directly by the net valence on plasma proteins (the Donnan effect). Kamel and colleagues12 showed that a rapid infusion of saline caused a significant decline in the net anionic charge on plasma proteins. Based on this reasoning, it should be potentially dangerous to give a bolus of saline to children with DKA unless there was a definite haemodynamic indication to do so (for example, low blood pressure). In our experience, haemodynamic collapse is not common in children with DKA. If there is a haemodynamic indication, up to 3 mmol NaCl per kg isotonic saline should be infused over 90 minutes. Once the blood pressure stabilises, the rate of infusion of NaCl should be much slower, with an upper limit reaching a positive balance of 6 mmol NaCl per kg over 24 hours to replace the likely deficit of Na+.13
Bolus of insulin
The Na+/H+ exchanger (NHE) in cell membranes is normally inactive because it catalyses an electroneutral exchange and its substrates have a much higher concentration (ECF Na+ and ICF H+) than its products (ICF Na+ and ECF H+) in steady state (fig 1, bottom portion). Following a large intravenous bolus of insulin, this NHE could become active.14,15 The other activator of NHE is a higher H+ concentration in the ICF compartment.14,15 When NHE is active, there should be a gain of Na+ and a loss of H+ in the ICF compartment; this will increase the number of solute molecules in the ICF16 because the bulk of the exported H+ were bound primarily to ICF proteins or entered cells along with β-hydroxybutyrate on the monocarboxylic acid transporter (fig 1).17–19 Since intracellular acidosis is usually present in patients with DKA and there is an insulin receptor in the brain,20 an intravenous bolus of insulin could have a more dramatic intracerebral effect if it were given early on, when the BBB might be less restrictive to the passage of insulin.8–10 Although large doses of insulin were given in the past to patients with a serious degree of DKA, the incidence of cerebral oedema was quite low. This might reflect a less aggressive infusion of saline and/or the administration of insulin by the subcutaneous route.
While NaHCO3 would lower the ECF H+ concentration, this should only become important for Na+ entry into brain cells when their NHE is activated. If true, a combination of a bolus of insulin together with NaHCO3 and administration of both early in time could make this “trio” become important risk factors. In contrast, if insulin and NaHCO3 were administered later when the BBB is more intact, they might be less likely to cause cerebral oedema.
RISK FACTORS FOR CEREBRAL OEDEMA LATER IN THERAPY FOR DKA
Rapid fall in Pglu
A rapid decline in PGlu would be a risk factor if there were a slower decline in the concentration of glucose and related metabolites in the brain.3,4,8,9,21 The actual importance of this risk factor is not clear because the major fall in PGlu occurs much earlier than the clinical onset of cerebral oedema.
Fall in effective Posm
A fall in PGlu would lead to a decline in the effective Posm if PNa did not rise by an appropriate amount (fig 2, right portion). The PGlu usually falls by 5 mmol/l/h over the initial six hours of therapy. If the PGlu on admission were close to 50 mmol/l, the decline in PGlu would be close to 30 mmol/l. To avoid a fall in the “effective” Posm, the PNa must rise by 15 mmol/l or half the fall in PGlu (30 mmol/l). This is not a problem when the PNa is close to 125 mmol/l with a blood sugar of 50 mmol/l, which is common in adults with DKA.22 Therefore the target for the PNa would be 140 mmol/l in an adult population. In contrast, because the initial PNa is often close to 140 mmol/l on admission in a paediatric patient with a PGlu close to 50 mmol/l,23–25 there is a therapeutic dilemma. If the goal of therapy were to maintain a constant effective Posm over the initial 12–24 hours, PNa should rise to close to 155 mmol/l in these patients. This value itself tends to make clinicians uncomfortable with the need to defend the effective Posm vigorously. Although limited data were available in our preliminary study,23 it seemed advisable to maintain a constant effective Posm, because the eight patients who did not develop cerebral oedema had an effective Posm of 341 ± 7 mOsm/kg H2O from the 7 to the 24 hour time (PNa rose to 156 ± 1 mmol/l). In contrast, the six patients who developed cerebral oedema during therapy had a fall in their effective Posm to 307 ± 7 mOsm/kg H2O over this period (PNa remained at 140 ± 1 mmol/l).
CONCLUDING REMARKS
Clinical management of DKA should change. Each mode of therapy should be administered aggressively only if it deals with a threat to life. The cautions listed below are most important in a paediatric population because they are at greater risk of developing cerebral oedema. A constant infusion rather than a bolus of insulin should be given as the only seconds to minutes emergency that is reversed by insulin is life threatening hyperkalaemia (see Halperin and colleagues26). Isotonic saline should be infused rapidly only if there is a significant degree of hypotension. An upper target for a positive Na+ balance is 6 mmol/kg in the first 24 hours. Isotonic saline is the preferred intravenous solution during the first day because the aim is to prevent a fall in the effective Posm. In our opinion, it is advisable to select an appropriate target for the rise in PNa to decrease the degree of fall in the effective Posm. One must be vigilant concerning both the infusions given and the clinical picture, because if the CNS status does not continue to improve, cerebral oedema should be suspected. In this setting, we would give a rapid infusion of hypertonic saline or mannitol before there is irreversible brain damage. One should not limit the impression of the danger of cerebral oedema to mortality or gross cerebral function loss because cerebral oedema could lead to a loss of subtle cerebral functions that might not be easily recognised. Finally, this discussion points out a potential difficulty in interpreting data from large multicentre studies unless timing of therapy, prior deficits of Na+ for instance (haemodynamic signs), and combinations of risk factors are included in the analysis.
REFERENCES
Footnotes
-
This study was supported by a grant from the Canadian Institutes for Health Research (FRN 15485)
Linked Articles
- Atoms
- Miscellanea