Article Text

Abstract
OBJECTIVE To establish criteria for the diagnosis of medium chain acyl-CoA dehydrogenase (MCAD) deficiency in the UK population using a method in which carnitine species eluted from blood spots are butylated and analysed by electrospray ionisation tandem mass spectrometry (ESI-MS/MS).
DESIGN Four groups were studied: (1) 35 children, aged 4 days to 16.2 years, with proven MCAD deficiency (mostly homozygous for the A985G mutation, none receiving carnitine supplements); (2) 2168 control children; (3) 482 neonates; and (4) 15 MCAD heterozygotes.
RESULTS All patients with MCAD deficiency had an octanoylcarnitine concentration ([C8–Cn]) > 0.38 μM and no accumulation of carnitine species > C10 or < C6. Among the patients with MCAD deficiency, the [C8–Cn] was significantly lower in children > 10 weeks old and in children with carnitine depletion (free carnitine < 20 μM). Neonatal blood spots from patients with MCAD deficiency had a [C8–Cn] > 1.5 μM, whereas in heterozygotes and other normal neonates the [C8–Cn] was < 1.0 μM. In contrast, the blood spot [C8–Cn] in eight of 27 patients with MCAD deficiency > 10 weeks old fell within the same range as five of 15 MCAD heterozygotes (0.38–1.0 μM). However, the free carnitine concentrations were reduced (< 20 μM) in the patients with MCAD deficiency but normal in the heterozygotes.
CONCLUSIONS Criteria for the diagnosis of MCAD deficiency using ESI-MS/MS must take account of age and carnitine depletion. If screening is undertaken at 7–10 days, the number of false positive and negative results should be negligible. Because there have been no instances of death or neurological damage following diagnosis of MCAD deficiency in our patient group, a strong case can be made for neonatal screening for MCAD deficiency in the UK.
-
Electrospray ionisation tandem mass spectrometry (ESI-MS/MS) of acylcarnitine species is a reliable first line test for the diagnosis of MCAD deficiency
-
Criteria for the diagnosis of MCAD deficiency using ESI-MS/MS must take account of age and carnitine depletion
-
If screening is undertaken at 7–10 days, the number of false positive and negative results should be negligible
-
Because there have been no instances of death or neurological damage following diagnosis of MCAD deficiency in our patient group, a strong case can be made for neonatal screening for MCAD deficiency in the UK
- Reye syndrome
- hypoglycaemia
- fatty acid oxidation
- blood spots
- acylcarnitines
Statistics from Altmetric.com
Inborn errors of fatty acid oxidation are an important cause of morbidity and mortality in the UK. The commonest disorder is medium chain acyl-CoA dehydrogenase (MCAD) deficiency and the commonest genotype responsible for the disorder is homozygosity for the A985G mutation in the MCAD gene. This genotype has a prevalence of one in 11 000 in the West Midlands and one in 28 000 in the Trent area.1-3 Children with MCAD deficiency are normal when examined and for most of the time they are asymptomatic; the diagnosis is usually only suspected when they present acutely with hypoglycaemia or a Reye-like illness, often precipitated by an infection, at a median age of between 10 and 14 months.4 5 Metabolic decompensation often requires a period of intensive care and is associated with a mortality of 24%.4 Among the survivors, 37% have neurodevelopmental problems.4 Once the diagnosis has been made, metabolic decompensation can be prevented by ensuring that the child receives regular high carbohydrate drinks (or occasionally intravenous dextrose) at times of infection or anorexia. We are aware of only one child with MCAD deficiency in the UK who has died once the diagnosis of MCAD deficiency has been made. Unfortunately, the traditional method for screening for inborn errors of intermediary metabolism (analysis of urinary organic acids by gas chromatography-mass spectrometry (GC-MS)) does not always detect any abnormality when a child with MCAD deficiency is well.
Pioneering work by Millington et al has led to the introduction of a new method for screening for disorders of fatty acid oxidation based on the analysis of carnitine species in dried blood spots (on a Guthrie card) using tandem mass spectrometry (TMS or MS/MS).6 7 The first TMS method to be validated for the diagnosis of MCAD deficiency involved conversion of the carnitine species to their methyl esters and ionisation by the liquid secondary ionisation technique (LSI-MS/MS). Using this method, Van Hoveet al established the following diagnostic criteria for MCAD deficiency: octanoylcarnitine (C8carnitine) concentration > 0.3 μM, a ratio of octanoylcarnitine:decanoylcarnitine (C10 carnitine) of > 5, and a lack of an increase of carnitine species > C10. Unaffected family members (including heterozygotes for MCAD deficiency) had C8 carnitine concentrations < 0.3 μM.7
It has proved difficult to automate sample injection with LSI-MS/MS, but this can be accomplished easily by means of an alternative method—electrospray ionisation tandem mass spectrometry (ESI-MS/MS).8 9 If the sample is subjected to butylation rather than methylation, accurate measurement of blood spot amino acid concentrations can be achieved in the same analysis. This paper describes our experience of diagnosing MCAD deficiency using ESI-MS/MS analysis of butylated carnitine species from a blood spot.
Methods
ELECTROSPRAY IONISATION TANDEM MASS SPECTROMETRY
Blood samples were collected on a standard neonatal screening Guthrie card. A 6 mm disc (equivalent to 10 μl of blood) was punched from the dried blood spot and the carnitine species eluted with 250 μl methanol containing deuterated internal standards in amounts equivalent to the following concentrations in blood: D3carnitine, 25 μM; D3 octanoylcarnitine, 1.0 μM; and D3 palmitoylcarnitine, 5.0 μM. During the early part of the study, the eluate was dried and heated with butanol/acetyl chloride to convert the carnitine species to the butyl esters.9Subsequently, it was shown that identical results were obtained by derivatising the sample at room temperature overnight. This procedure has advantages for the automation of the process and was subsequently adopted for all samples. The reaction products were reconstituted in 60 μl acetonitrile water (60/40 vol/vol), and 5 μl was injected from an autosampler into the electrospray ionisation source of a Quattro I tandem mass spectrometer (Micromass, Altringham, UK), operating in positive ion mode. The capillary voltage was 3.65 kV, cone was voltage 27 V, collision energy was 35 eV, and argon was used as the collision gas at a pressure of 2.3 × 10−3 mBarr. Free and acyl carnitines were detected and measured by setting the instrument to detect precursors of m/z (mass/charge ratio) 85.2+— this ion is produced by the fragmentation, in the collision cell, of butyl carnitine and butylated acyl carnitines. The mass range from m/z 210 to 550 was scanned for 24 seconds (six scans). Raw mass spectra were subjected to background subtraction, smoothing, and centroid conversion using MassLynx software (Micromass). All the carnitine profiles generated were inspected visually. The NeoLynx program was used to compute ratios of peak intensities. The ratio, intensity of m/z 344.2 (octanoylcarnitine)/intensity of m/z 347.2 (D3octanoylcarnitine), gives the blood spot octanoylcarnitine concentration (μmol/l), and the ratio m/z 344.2:372.2 reflects the octanoylcarnitine:decanoylcarnitine ratio in the sample.7In addition, the ratios of m/z 218.2:221.2 (free carnitine:D3 carnitine), and m/z 456.3:459.3 (palmitoylcarnitine:D3 palmitoylcarnitine) were used to calculate the concentrations of free carnitine and palmitoylcarnitine in the blood sample.
BLOOD SPOT DNA ANALYSIS
The A985G mutation in exon 11 of the MCAD gene introduces an NcoI restriction site into the mutant but not the normal allele. Thus, the mutant allele can be distinguished from the normal allele by NcoI digestion of DNA obtained from a polymerase chain reaction (PCR), followed by agarose gel separation of the products. The original method described by Yokota et al has been modified to include a restriction enzyme site within the antisense primer to determine the efficiency of the enzyme digestion and remove false negative results. Digestion with NcoI of the 209 bp (base pairs) PCR product gives two DNA fragments of 178 bp and 31 bp in the normal allele, but three fragments of 20 bp, 158 bp, and the control 31 bp fragment in the mutant. The 178 bp and 158 bp fragments can be resolved to allow identification of mutant alleles.
PCR amplification of genomic DNA from 1 mm2 blood spots was performed with the following primers: sense, 5′-TTTATGCTGGCTGAAATGGCCATG; antisense, 5′-AGTTTTTCTACAGGATATTCTGTATTAAATCCATGGCCTC. The NcoI sites are shown in bold, including the partial site within the sense primer.
DNA was amplified in a mixture (50 μl) containing 160 mM (NH4)2SO4, 670 mM Tris-HCl (pH 8.8), 1.5 mM MgCl2, 0.1% Tween-20, 2 mM dNTPs, oligonucleotide primers as indicated above (20 pmol each), and 2 U BIOPROTH polymerase (Bioline, London, UK). Amplification was performed in a Omnigene cycler (Hybaid, Middlesex, UK) as follows: seven minutes at 94°C before cycling, then 35 cycles of one minute at 94°C, one minute at 59.5°C, one minute at 74°C, followed by seven minutes at 74°C.
DNA was precipitated with 2.5 volumes of 100% ethanol, and resuspended with 50 mM Tris-HCl (pH 8.0), 10 mM MgCl2, 100 mM NaCl, 5 mM spermidine trihydrochloride, 1 U NcoI, and deionised water to a final volume of 20 μl. After two hours, digested DNA was analysed on a 2.5% nusieve agarose gel (Sigma, Poole, Dorset, UK).
PATIENT SAMPLES
The protocol for analysis of samples from patients with MCAD deficiency and from controls was approved by the ethics committee of Great Ormond Street Hospital.
During the period September 1993 to September 1997, all children seen by members of the Metabolic Unit at Great Ormond Street Hospital with clinical features that could be caused by a fatty acid oxidation defect or organic acidaemia had a Guthrie card sent for analysis of carnitine species. Staff on the intensive care unit were also encouraged to send samples from all new admissions. In addition, cards were sent from children known to have MCAD deficiency attending the Metabolic Clinic. None of these children were receiving carnitine supplements. Their treatment regimen involved avoidance of fasting and the use of an “emergency regimen”.10 Blood samples were also obtained from parents and siblings of children known to have MCAD deficiency and in one case from the daughter of a known A985G homozygote. Paediatricians telephoning for advice on children with clinical features that could be caused by a fatty acid oxidation defect or organic acidaemia were encouraged to send blood spots for analysis and from September 1995 onwards, many unsolicited samples were received. When we were notified by a pathologist of a case where there was fat accumulation in the liver, muscle, heart, or kidney at postmortem examination, we contacted the regional screening centre to obtain the unused spots from the blood that had been obtained at 7–10 days for phenylketonuria/hypothyroidism screening. Neonatal blood spots were also retrieved from patients known to have MCAD deficiency. To establish normal ranges for the neonatal blood spots, 482 unused blood spots were obtained from the North Thames screening laboratory. These blood spots will be referred to as neonatal screening samples as opposed to the remainder, which will be referred to as selective screening samples (n = 2168).
The independent tests that were used or had been used to diagnose or exclude MCAD deficiency in this cohort of patients collected between 1993 and 1997 were as follows: plasma non-esterified fatty acid:3-hydroxybutyrate ratio at the time of an episode of hypoglycaemia (occasionally provoked by a diagnostic fast),11 gas chromatography-mass spectrometry analysis of urine organic acids following such an episode (looking specifically the presence of hexanoylglycine and suberylglycine), phenylpropionic acid load,12 blood spotcis-4-decenoic acid,13 and white cell/fibroblast tritium release assay (using3H-myristate/3H-oleate, and3H-octanoate).
MANAGEMENT OF PATIENTS
Following the diagnosis of MCAD deficiency, the parents, general practitioner, and paediatrician were given careful advice on the importance of avoidance of fasting and the use of regular high carbohydrate drinks during periods of infection and anorexia.10 To avoid delays in the casualty department, paediatricians were asked to give patients free access to the children’s ward so that there was no delay in starting a dextrose infusion if the child was vomiting the emergency regimen drinks.
STATISTICAL ANALYSIS
The analysis of distributions of analyte concentrations in blood spots was performed with software from Statistics Package for the Social Sciences (SPSS). If two sets of values showed a normal distribution and an F test showed that the values had the same variance, the Student’s t test was used to determine the significance of the difference between the two distributions; if not, the Mann-Whitney test was used. A value of p < 0.05 was regarded as significant.
Results
OCTANOYLCARNITINE CONCENTRATION IN SELECTIVE SCREENING BLOOD SPOTS
Visual inspection of most of the selective screening blood spots (2075 samples) showed a normal profile; the only carnitine species present (apart from the internal standards) were free carnitine (m/z 218), “acetylcarnitine” (m/z 260), palmitoylcarnitine (m/z 456), and oleoylcarnitine (m/z 482).8 (It has been shown previously that glutamate in the blood spot can contribute to the m/z 260 peak when butyl esters of acylcarnitines and amino acids are analysed.8 9) There was no octanoylcarnitine peak (indicating that the concentration of octanoylcarnitine was < 0.1 μM). The mean (SD) concentrations of free carnitine and palmitoylcarnitine in these blood spots were 34.7 (10.8) μM and 1.98 (0.55) μM, respectively. One hundred and thirty eight of the selective screening blood spots had detectable octanoylcarnitine; 65 of these had peaks attributable to other acyl carnitines not normally detected including C4 (m/z 288), C5 (m/z 302), C12 (m/z 400), and C14 (m/z 428). This profile is not compatible with MCAD deficiency. Some of these patients had multiple acyl-CoA dehydrogenase deficiency, some ketotic hypoglycaemia, and others had severe illness/multi-organ failure; the full range of conditions that can give rise to this profile will be considered elsewhere. In 27 of the selective screening samples, a small octanoylcarnitine peak was seen but the ratio to the D3 octanoylcarnitine peak indicated a concentration less than 0.38 μM. This group included some children and adults who were heterozygous for the A985G or C250T mutation (see below); none of the patients with an octanoylcarnitine concentration < 0.38 μM had evidence of MCAD deficiency by other laboratory criteria. In fig 1 the dotted line is drawn at 0.38 μM for samples obtained after 2 weeks of age to indicate that this is the upper limit of the normal range for this age group.
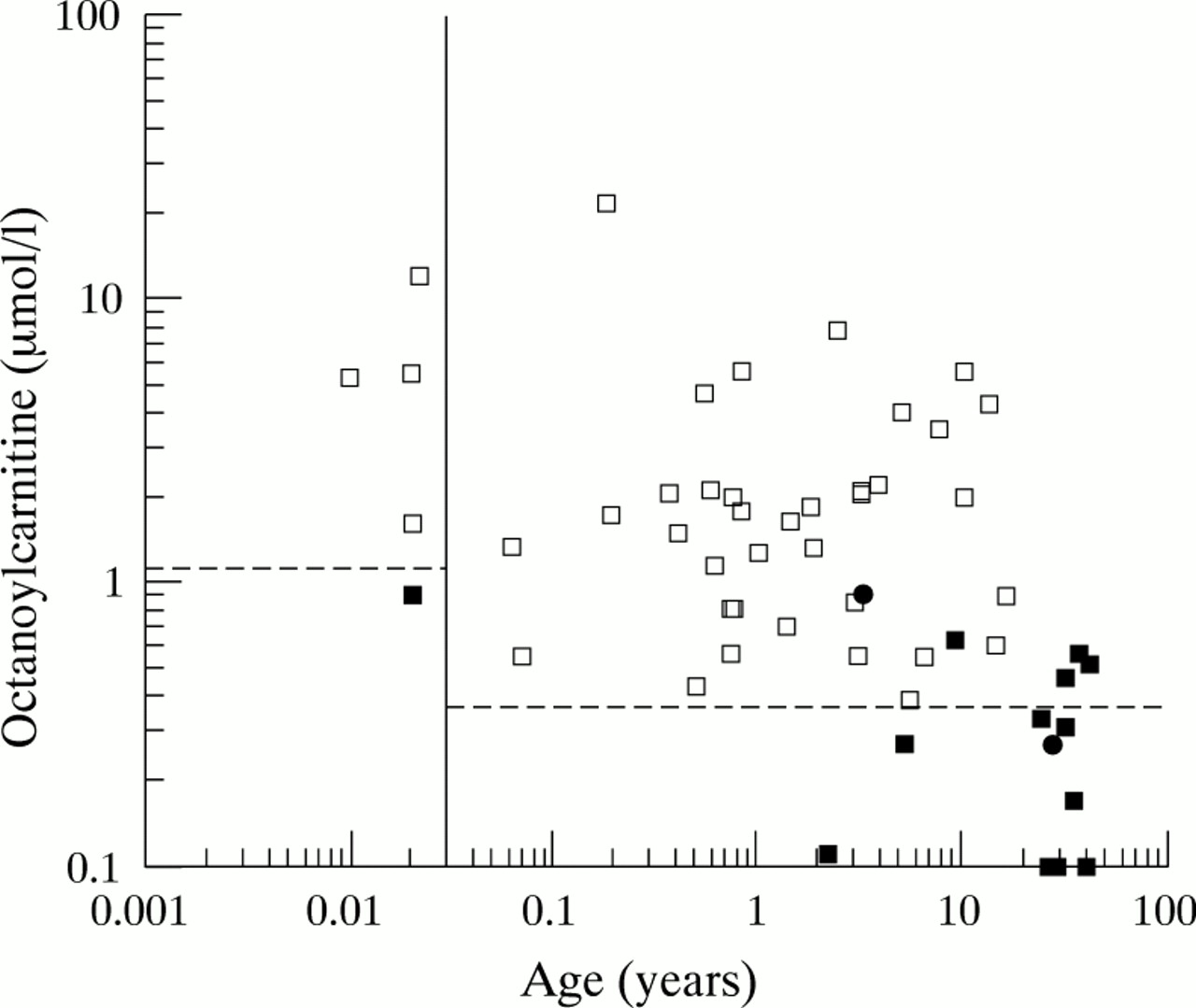
{kind=link}
Age distribution of blood spot octanoylcarnitine concentrations in patients with MCAD deficiency (open squares), A985G heterozygotes (filled squares), C250T heterozygotes (filled circles), and controls (all values below dotted line).
OCTANOYLCARNITINE CONCENTRATION IN NEONATAL SCREENING BLOOD SPOTS (CONTROLS)
Visual inspection of the spectra from the 482 neonatal blood spots suggested that a significant proportion had a detectable octanoylcarnitine peak. The NeoLynx software was used to generate a value for the octanoylcarnitine concentration for each sample in which an m/z 344.2 peak was detectable above the background noise. Statistical analysis of these results revealed a bimodal distribution—detectable and undetectable C8 carnitine—and a skewed distribution among infants with detectable C8carnitine. Taking the sample as a whole, 75% of values were below 0.4 μM, the range was from 0 to 0.82 μM, and there were five outliers with a blood spot octanoylcarnitine concentration between 0.82 and 1.0 μM. DNA testing was not performed on these samples, but there is a possibility that they represent samples from neonates who are heterozygotes for MCAD deficiency (see below), which occurs at a frequency of at least one in 50 to one in 80 in UK populations.1-3 In fig 1, the dotted line (upper limit of normal) is drawn at 1.0 μM for blood samples obtained before 2 weeks of age (0.04 years). The mean (SD) concentration of free carnitine in the neonatal blood spots was 35.8 (11.7) μM, which was not significantly different to the selective screening samples.
BLOOD SPOTS WITH AN OCTANOYLCARNITINE CONCENTRATION SUGGESTIVE OF MCAD DEFICIENCY
Forty three selective screening blood spots were identified in which the octanoylcarnitine concentration was > 0.38 μM and in which, apart from the C8, there was only an increase in C6 and/or C10 carnitine. This group contained all the samples from 14 patients already known to have MCAD deficiency on the basis of conventional tests (organic acid analysis,cis-4-decenoic acid analysis, A985G homozygosity, phenylpropionic acid load) and all of the samples from children subsequently proven to have MCAD deficiency on this basis (20 patients). The group also contained one child who had been classified erroneously as having multiple acyl-CoA dehydrogenase deficiency on the basis of organic acid analysis and a tritium release assay; she had the typical blood spot acylcarnitine profile of MCAD deficiency and her DNA analysis showed her to be a A985G homozygote. The values for the blood spot octanoylcarnitine concentration in 40 samples from the 35 patients with MCAD deficiency are shown in fig 1. There were five MCAD heterozygotes who had a blood spot octanoylcarnitine concentration > 0.38 μM (see below and fig 1). The earliest sample obtained from each patient with MCAD deficiency was used for more detailed analysis of the determinants of the octanoylcarnitine concentration. The mean octanoylcarnitine concentration in the 35 patients with MCAD deficiency was 3.04 μM (range, 0.39–21.8).
DETERMINANTS OF OCTANOYLCARNITINE CONCENTRATION IN PATIENTS WITH MCAD DEFICIENCY
Age
The octanoylcarnitine concentration was found to be age dependent (fig 1). All four samples obtained before 2 weeks of age from neonates with MCAD deficiency had an octanoylcarnitine concentration above 1.5 μM. The eight infants less than 10 weeks old had a mean (SEM) octanoylcarnitine concentration of 6.9 (2.6) μM—significantly higher (p < 0.05) than the value seen in older infants and children (1.7 (0.3) μM). The concentration of free carnitine was also higher in the young infants with MCAD deficiency, but this difference was not significant. The mean (SEM) concentration of palmitoylcarnitine in the blood spots from neonates under 2 weeks of age with MCAD deficiency was 2.1 (0.3) μM—significantly higher than the mean (SEM) concentration of palmitoylcarnitine in older infants and children with MCAD deficiency (1.0 (0.1) μM). The mean concentration of palmitoylcarnitine in the children with MCAD deficiency who were older than 2 weeks was also significantly lower than the mean (SEM) palmitoylcarnitine concentration in the normal controls (1.98 (0.01) μM).
Carnitine depletion
The relation between carnitine depletion (as indicated by low blood spot free carnitine) and the concentration of octanoylcarnitine in the blood spot was investigated further. Children with a free carnitine concentration < 20 μM had a mean (SEM) octanoylcarnitine concentration of 1.12 (0.17) μM, whereas children with a normal concentration of free carnitine had an mean (SEM) octanoylcarnitine concentration of 4.3 (1.1) μM, which was significantly higher (p < 0.05).
Fasting state
The effect of fasting on the blood spot octanoylcarnitine concentration was determined in two patients who required a fasting test for the purposes of advising parents on management. The concentration after an overnight fast in a 16 month old infant was 0.56 μM; after a 20 hour fast the value was 0.93 μM. A rise from 1.4 to 1.8 μM was seen in a 6½ year old.
Genotype
The A985G mutation was detected in 58 out of the 66 alleles in which it was sought. Twenty six of the 33 patients were homozygous for the A985G mutation. Six had the A985G mutation on one chromosome but not on the other; all had an organic acid profile when stressed, which contained the metabolites characteristic of MCAD deficiency, including suberylglycine and hexanoylglycine. One of these five compound heterozygotes patients was shown to have the genotype A985G/C250T (Dr B Andresen, personal communication). The remainder of the mutant alleles have yet to be determined. One patient did not have the A985G mutation on either chromosome but had an organic acid profile at the time of her acute presentation that was diagnostic of MCAD deficiency. There were no differences in the octanoylcarnitine concentrations between A985G homozygotes and other patients with MCAD deficiency.
THE OCTANOYLCARNITINE:DECANOYLCARNITINE RATIO IN PATIENTS WITH MCAD DEFICIENCY
The ratio of m/z 344 (octanoylcarnitine) to m/z 372 (decanoylcarnitine) was extremely variable in the MCAD samples. The mean value was 6.6. The lowest value recorded was 0.82 in a 9 month old infant, whose blood sample was obtained at the time of presentation with acute encephalopathy and who had a very low free carnitine concentration. The highest measurable ratio was 31 and there were three children in whom the ratio could not be measured because the decanoylcarnitine concentration was below the limits of detection of the method (< 0.1 μM). These children with a high C8:C10 ratio all had normal free carnitine concentrations.
BLOOD SPOT OCTANOYLCARNITINE CONCENTRATIONS IN HETEROZYGOTES FOR MCAD DEFICIENCY
Blood spot acylcarnitine concentrations were determined in: (1) 14 parents and siblings of the A985G homozygous children (and of the C250T/A985G compound heterozygote) and (2) the daughter of an adult who was homozygous for the A985G mutation, after they had been shown to carry only one mutant allele. The children heterozygous for the A985G mutation had the following blood spot octanoylcarnitine concentrations: 0.90 μM in a neonate, 0.11 μM in a girl of 2¼ years, 0.27 μM in a girl of 5¼ years, and 0.62 μM in a boy of 9 years. A 3 year old girl who was heterozygous for the C250T mutation had a blood spot octanoylcarnitine concentration of 0.92 μM. Adult heterozygotes had blood spot octanoylcarnitine concentrations between undetectable (< 0.1 μM) and 0.56 μM. The blood spot octanoylcarnitine concentrations seen in the heterozygotes are included in fig 1. The free carnitine concentrations in children with octanoylcarnitine concentrations between 0.35 and 1.0 μM, who were homozygous for MCAD deficiency were compared with those found in the heterozygotes with this range of octanoylcarnitine concentrations. Whereas all the homozygous patients with MCAD deficiency had a free carnitine concentration less than 20 μM (mean (SD), 11.7 (5.4) μM), in the heterozygotes there was no such evidence of carnitine depletion; the mean (SD) free carnitine concentration was 33.7 (7.9) μM (not significantly different to the controls).
There was one blood spot that showed an octanoylcarnitine concentration > 0.38 μM from a child who did not carry the A985G mutation on either of her chromosomes. Insufficient information was available to exclude MCAD deficiency. However, a modestly raised blood spot octanoylcarnitine concentration (0.51 μM) associated with a normal free carnitine concentration (35 μM) would be compatible with heterozygosity for a mutation other than A985G.
OUTCOME
The outcome for this group of patients with MCAD deficiency is still being documented. One patient died before a diagnosis was made, the diagnosis being made on acylcarnitine and genotype analysis of a retrieved neonatal blood spot. Five of the 37 patients had older siblings who died. None of the patients in this cohort has died since the diagnosis was made. We are aware of three episodes of admission to hospital to cover episodes of vomiting; many more episodes of infection have been treated by the parents and general practitioners at home.
Discussion
METHODOLOGY
Our study showed that the measurement of octanoylcarnitine by electrospray ionisation TMS of butylated carnitine species in patients with MCAD deficiency and in controls produces results that are similar to those reported by Van Hove et al using liquid secondary ionisation TMS analysis of methyl esters. The electrospray method has the substantial advantage that, with automatic injection from microtitre plates, it is possible for an unattended instrument to analyse amino acids and carnitine species from 600 blood spots each day. The front source of the mass spectrometer needs to be cleaned every third day. Currently, the pipetting steps in the elution and derivatisation steps are still performed by hand in our laboratory but, particularly with room temperature butylation, this process is likely to be automated soon.
DETERMINANTS OF BLOOD SPOT OCTANOYLCARNITINE CONCENTRATION IN PATIENTS WITH MCAD DEFICIENCY
Our study has confirmed that the most important criterion for the detection of MCAD deficiency is the presence in the blood spot of octanoylcarnitine at a concentration > 0.3 μM (in this study > 0.38 μM). However, we have also shown that blood spot octanoylcarnitine concentrations are higher in neonates with MCAD deficiency and that there is an association between low octanoylcarnitine and low free carnitine. There are two possible explanations for the latter association. The first is that the volume of blood in the 6 mm disc was substantially less than 10 μl or that the elution was much less efficient for this group of blood spots. We consider this to be very unlikely; all the Guthrie cards that were received were made from approved brands of filter paper, all blood spots were inspected visually to make sure that the 6 mm disc was completely filled, and a standardised procedure was adopted for the elution step. The second and more likely explanation for the association is that these patients were carnitine depleted at the time the sample was taken. It is likely that placental transfer of carnitine ensures that patients with MCAD deficiency do not become carnitine deficient in utero. Breast milk contains 50 μM carnitine and it is thought that this might be sufficient to prevent carnitine depletion in the first few months of life in breast fed infants with MCAD deficiency.14 After weaning, a child with MCAD deficiency on a normal diet will often have a total carnitine concentration in plasma that is 10–50% of the normal concentration.4Carnitine given orally at a dose of 100 mg/kg leads to a large rise in the urinary excretion of octanoylcarnitine.14 Thus, the most plausible explanation for the low plasma octanoylcarnitine concentration seen in some of our infants aged over 6 months is carnitine depletion. It is important to recognise that this carnitine depletion must be taken into consideration when making the diagnosis of MCAD deficiency after 6 months of age (and perhaps earlier if the infant is not breast fed or receiving a carnitine supplemented formula). In contrast to our results, Van Hove et al did not find a significant difference between blood spot octanoylcarnitine concentrations in children with MCAD deficiency who were receiving an L-carnitine supplement and those who were not.7 The reason for this is not immediately clear, particularly as the same group found that L-carnitine corrects the low plasma free carnitine concentration and increases the urinary octanoylcarnitine excretion.14 The effects of carnitine supplementation require further study.
Sequential analyses of octanoylcarnitine concentrations in two MCAD patients during a fast suggested that the octanoylcarnitine concentration is only 25–70% higher after 20 hours fasting than after 16 hours fasting. Thus, we have found no evidence that the time of sampling in relation to the last meal has a profound effect on the blood spot octanoylcarnitine result.
BLOOD SPOT OCTANOYLCARNITINE CONCENTRATIONS IN MCAD HETEROZYGOTES
The study by Van Hove et al found that all heterozygotes for MCAD deficiency had blood spot octanoylcarnitine concentrations less than 0.3 μM.7 In our study, which uses a slightly different methodology, seven of the 15 heterozygotes had values less than 0.3 μM and nine had values within our normal range (< 0.38 μM). However, there were six heterozygotes who had a blood spot octanoylcarnitine of between 0.38 and 1.0 μM—that is, similar to the values seen in carnitine depleted homozygotes. These heterozygotes had a normal free carnitine concentration in their blood spots. The highest octanoylcarnitine concentration in our group of heterozygotes was seen in the C250T heterozygote, but most of our heterozygotes had a single copy of the A985G mutation (as was the case with the Van Hove et al series). The reason(s) for the discrepancy between our results and those of Van Hove et al are not immediately clear. There are significant differences between our respective methods and the relevant measurements of octanoylcarnitine are fairly close to the limits of sensitivity of the first generation of tandem mass spectrometers. However, these factors would not be expected to have a great effect on an isotope ratio based measurement, nor would they be expected to produce an artefactual increase in octanoylcarnitines in heterozygotes or other normal individuals. It is possible that there is a common polymorphism in UK patients, which is not so prevalent in the USA, and that the presence of this polymorphism on the normal chromosome gives rise to an increased octanoylcarnitine concentration in the same way as the G625A polymorphism in the short chain acyl-CoA dehydrogenase gene gives rise to ethylmalonic aciduria.15We do not believe that six of 15 first degree relatives of known MCAD homozygotes (mostly A985G/A985G) can be compound heterozygotes, because this would require that there should be an MCAD causing mutation that is more prevalent than A985G, and no such mutation has ever been detected. It is possible that there are differences between the UK and USA heterozygotes in terms of feeding/fasting patterns, or in terms of dietary intake of carnitine, long chain triglyceride, or medium chain triglyceride. Clearly, further research is required.
The neonatal screening programme in Pennsylvania produced an unexpectedly high incidence of MCAD deficiency and an unexpectedly low percentage of A985G homozygotes.16 If a pilot neonatal screening programme in the UK produces similar results it will be necessary to prove that the individuals identified are not heterozygotes.
DIAGNOSTIC CRITERIA FOR MCAD DEFICIENCY
Based on the experience of our study, we propose the following criteria for diagnosis of MCAD deficiency when butylated carnitine species from a blood spot are analysed by ESI-MS/MS analysis (parents of m/z 85.2+): for routine neonatal blood spots (4 days to 2 weeks), an octanoylcarnitine concentration > 1.2 μM and no increase in carnitine species < C6 or > C10. For blood spots obtained later than this, an octanoylcarnitine concentration > 1.0 μM, or an octanoylcarnitine concentration between 0.38 and 1.0 μM coupled with evidence of carnitine depletion (free carnitine < 20 μM) and, in both cases, no increase in carnitine species < C6 or > C10. Because it is easy to check for the A985G mutation on a 1 mm2 portion of a blood spot, this should be done on all samples that produce a positive diagnosis on TMS screening. In cases where the blood spot octanoylcarnitine is < 1.0 μM and DNA analysis does not reveal homozygosity for the A985G mutation, further tests are required to determine whether the child is a heterozygote or a compound heterozygote (that is, affected by MCAD deficiency). A useful test in this context will be the blood spotcis-4-decenoic acid concentration.13
The data obtained in our study suggest that the diagnostic criterion proposed by Van Hove and colleagues7 of an octanoylcarnitine:decanoylcarnitine ratio of > 5 is not applicable to patients with severe carnitine depletion and/or patient samples analysed by ESI-MS/MS of butylated carnitine species rather than LSI-MS/MS of methyl esters.
DETERMINANTS OF OUTCOME IN MCAD DEFICIENCY
None of our patients with MCAD deficiency has died once the diagnosis has been made. We believe that this is because the parents, general practitioners, and local paediatricians know how to avoid metabolic decompensation when the child develops an intercurrent infection such as gastroenteritis. Support for this view comes from the USA experience of neonatal screening for MCAD deficiency. Out of 12 infants identified by a neonatal screening programme there were two deaths.16 In one case, the family could not get hold of their primary care physician quickly enough, in the second, the importance of the child’s MCAD deficiency was not fully appreciated by the physicians in the accident and emergency department when the child became ill away from home (E Naylor, personal communication, 1997). These problems in delivery of primary health care are now being addressed and, in the Pennsylvania programme, 20 cases have now been diagnosed and there have been no further deaths.
In our view, the case for carnitine supplementation is not yet established. We know from our experience of true systemic carnitine deficiency (carnitine transport defect) that low amounts of carnitine in the mitochondria of the liver, skeletal muscle, and cardiac muscle are associated with impaired function.4 The fact that administration of L-carnitine to a patient with MCAD deficiency leads to increased excretion of octanoylcarnitine suggests that there is accumulation of octanoyl-CoA in the mitochondria of these patients and that this can be reversed by carnitine treatment. This should free the sequestered CoA and enhance the CoA dependent reactions in the mitochondrion. On the other hand, one of the striking features of the carnitine profiles of the patients with MCAD deficiency in our study was the fact that most of these children had low blood spot concentrations of palmitoylcarnitine and oleoylcarnintine. This raises the possibility that, in MCAD deficiency, carnitine depletion leads to impaired formation of long chain acylcarnitines and that this limits the entry of long chain acyl-CoA into the mitochondrion and limits the β oxidation flux. This might provide some protection against acute metabolic decompensation. Further studies are required on the effects of carnitine supplementation in MCAD deficiency.
Conclusion
Electrospray ionisation tandem mass spectrometry has greatly simplified the diagnosis of MCAD deficiency (as well as other fat oxidation defects) in symptomatic children in our unit. The ease of obtaining the blood sample and sending it in the post means that the test can be performed easily by local paediatricians and even by primary care physicians. However, the only reliable way of reducing the risk of death and handicap, which often occurs at the time of the first decompensation (before the diagnosis has been made), is by making the diagnosis soon after birth by a neonatal screening programme. Our data suggest that tandem mass spectrometry will provide a reliable first line test for MCAD deficiency in neonatal blood spots. Screening for phenylketonuria by TMS can be done on the same butylated blood spot extract.
Acknowledgments
This project was funded by the Engineering and Physical Sciences Research Council via the LINK scheme. We also gratefully acknowledge generous donations (for the purchase of equipment) from the Foundation for the Study of Infant Deaths, the Madeleine Curtis Trust, and Allders plc.