
Abstract
We have developed a technique to screen for gross deletions/duplications and point mutations using one streamlined approach. Fluorescent multiplex quantitative PCR is used to determine the copy number of each exon, followed by conformation sensitive capillary electrophoresis (CSCE) of the same PCR products on a multi-capillary genetic analyser. We have developed this technique to screen all 79 exons of one of the largest human genes currently known (dystrophin) using 12 multiplex PCR assays. A blind trial of 50 male and 50 female samples, in which 84 mutations had previously been found and characterized by other techniques, showed 100% sensitivity and specificity. We then applied this method to screen over 100 patient samples previously screened for deletions and duplications of 28 exons from the two hotspot regions. Our data show that combining a full deletion/duplication screen with CSCE will detect a mutation in 98% of Duchenne muscular dystrophy patients and 93% of Becker muscular dystrophy patients where the clinical diagnosis is certain. This technique is applicable to any gene and is particularly suited to mutation screening of large genes, decreasing the time taken for a complete gene screen for nearly all mutation types.
Similar content being viewed by others

Introduction
Identification of pathogenic mutations in patients affected by Mendelian diseases confirms a clinical diagnosis and allows definitive carrier testing and prenatal diagnosis for family members. Precise knowledge of the mutation is required for some of the potential therapies currently being developed for genetic diseases, such as exon skipping,1 or for therapies involving suppression of premature termination codons.2 In families where no sample is available from an affected patient mutation screening can be applied to at-risk relatives. In the absence of identifying a mutation, a carrier risk can then be modified using Bayesian calculations; however, a not-insignificant risk will remain unless a method with high sensitivity is used. Xp21 muscular dystrophy is a classic example of a disease for which accurate determination of carrier status is complex; samples are not available from affected males in approximately 20% of families3 and the high rate of new mutation adds to the problem.4
Duchenne muscular dystrophy (DMD; OMIM 310200) is the most common lethal X-linked recessive disorder, affecting one in 3500 live male births.5 Becker muscular dystrophy (BMD; OMIM 300376) is less common, affecting one in 18 500 live male births.6 Both diseases are caused by mutations in the 2.4 Mb DMD gene at Xp21.
Approximately 70% of mutations causing DMD are large deletions or duplications of one or more exons, with the remaining 30% believed to be small insertion/deletion mutations and point mutations.7 The mutation spectrum for BMD is slightly different, with up to 85% of mutations believed to be large deletions or duplications and the remainder missense mutations or mutations affecting splicing.7
Mutation screening strategies for the DMD gene have tended to concentrate on screening for large deletions or duplications by methods such as Southern blotting,8 multiplex PCR,9, 10, 11 QF-PCR,12 MAPH13 and MLPA.14 Screening for nonsense mutations has been possible using the protein truncation test,15 but this is a time-consuming method, which is only practical if a muscle biopsy from an affected male is available. More recently, other methods have been described to screen the DMD gene for point mutations, including dHPLC,16 DOVAM,17 SCAIP,18 DGGE19 and complete sequencing of the 14 kb DMD cDNA.20 All of the DNA-based methods described so far screen either for large deletions and duplications, or for point mutations, but not both. RNA-based methods are capable of detecting all types of mutation but do not detect all gross duplications, and as mentioned for PTT, their major disadvantage is the need for fresh muscle from an affected male, which is often not available. Thus, no single method is available for detecting all mutations in affected males and carrier females. This dependence on a two-tiered (or more) approach to mutation screening applies to all genes. A unified method that utilized the same products for identifying all mutation types would logically be more efficient, cheaper and faster. We therefore developed a genomic DNA-based method to screen for deletions, duplications and point mutations throughout the entire coding region of the DMD gene in a single analysis.
To provide a simultaneous screen of the DMD gene for large deletions, duplications and point mutations, we combined QF-PCR for all 79 exons of the DMD gene (plus controls) with fluorescent multiplex conformation sensitive capillary electrophoresis (FM-CSCE) analysis of the same products, followed by sequencing of PCR fragments showing altered morphologies. The FM-CSCE assay was optimized, evaluated and subsequently validated in a blind trial. We then applied this screening method to 58 individuals with DMD and 51 with BMD. This method is applicable to both affected males and female carriers.
Materials and methods
DNA samples
DNA was obtained from the diagnostic DNA bank at Guy's Hospital. The DNA had been extracted from blood using a variety of different extraction protocols in different referring laboratories. For the assay development and optimization, all available samples with known DMD point mutations were selected. For the blind trial, a combination of male and female samples with whole exon deletions or duplications, point mutations, or no mutation in the DMD gene were selected and coded, so that samples could be identified only in retrospect, once the blind trial had been completed. Once the blind trial had been completed, the technique was used to screen DNA samples from a group of over 100 males with diagnoses of either DMD or BMD in whom neither deletion nor duplication had been detected when screening with the QF-PCR method described by Yau et al.12
DNA concentrations were measured for patients and controls and adjusted to 25 ng/μl for female samples and 50 ng/μl for male samples.
PCR primers
Primers were designed to amplify all 79 exons of the DMD gene (the entire 3′UTR is not covered), two alternative promoters (purkinje and cortical) and two exons of the myelin protein zero gene (MPZ) located at 1q22 to control for whole gene deletions or duplications. In all cases, primers were designed to amplify at least 50 bp either side of the exonic sequence for maximum efficiency in detecting mutations. One primer from each pair was 5′ labelled with FAM, VIC, NED or PET fluorophores (Applied Biosystems, Foster City, CA, USA). The other primer of the pair was not labelled and an unlabelled version of each labelled primer was also synthesized for use in sequence analysis (Invitrogen Corp., Carlsbad, CA, USA). Amplicon sizes ranged from 200 to 500 bp. The fragments were arranged into 12 multiplexes, each containing seven fragments. Two alternate sets of exon 63 primers were included as this exon occasionally showed inconsistent amplification in normal control samples. Primer and multiplex details are available in the Supplementary Information.
PCR conditions
PCR assays were set up to amplify all fragments simultaneously in 12 multiplexes in 96-well plates with eight patient samples per plate. PCR was carried out in a 25 μl volume containing 4 mM MgCl2 (2.5 mM in multiplex 8), 16 mM (NH4)2SO4, 67 mM Tris-HCl pH 8.3, 0.01% Tween-20, 0.5 mM each dNTP, 5 or 10 pmol each primer (see multiplex details in Supplementary Information), 2.5 U Immolase DNA polymerase (Bioline, London, UK) and 5 μl of diluted genomic DNA. Multiplex 11 also contained 2.5 μg BSA.
PCR assays were cycled as follows: 95°C 7 min (hot start), followed by 95°C 30 s, 55°C 30 s, 72°C 2 min for 22 cycles. Four microlitres of PCR product was removed for QF-PCR analysis after 22 cycles and the remaining products returned to the cycler for a further eight cycles of PCR.
Heteroduplex formation
Normal control male DNA was amplified for 30 cycles using the conditions described above with labelled PCR primers. Five microlitres of patient PCR product was mixed with 5 μl of PCR product from the normal male sample for all 12 multiplexes and denatured at 95°C for 11 min, followed by 23 1.5°C reductions in temperature at 1 min intervals. The mixing of patient samples with normal control DNA is only necessary for heteroduplex formation when the mutations being screened for are present hemizygously (eg in the case of a male DMD patient), or could be present homozygously (eg if applied to autosomal recessive disorders).
Capillary electrophoresis
The Applied Biosystems 3100 genetic analyser was used for QF-PCR, CSCE and sequencing throughout this study, with changes of polymer for the different applications accordingly.
FM QF-PCR
Four microlitres of PCR product was removed after 22 cycles of PCR and mixed with 0.3 μl GS-500 Liz size standard (Applied Biosystems), 15.7 μl Hi-Di Formamide (Applied Biosystems) and denatured at 95°C for 2 min. The products were electrophoresed on the Applied Biosystems 3100 using a 50 cm capillary, POP-6 polymer, standard electrophoresis module Genescan36_POP4 with the run time increased from 1500 to 5700 s.
CSCE
Heteroduplexed products were diluted 1:20 in 10 mM Tris-HCl. Two microlitres of diluted product was mixed with 0.3 μl GS-500 Liz size standard, 0.2 μl Immolase DNA polymerase PCR buffer (Bioline) and 17.5 μl 10 mM Tris-HCl. The products were electrophoresed on a 50 cm capillary array with polymer comprising 5.25% native Genescan polymer, 10% glycerol (Sigma), 5% Hi-Di Formamide and 1 × 3100 Genetic Analyser running buffer with EDTA. Run module Genescan36_POP4 was used with the following adaptations: temperature 18°C, voltage 10 kV, run time 4200 s.
Analysis
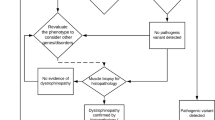
Data were analysed using Genescan and Genotyper software v3.7 (Applied Biosystems). QF-PCR results were analysed as described by Yau et al,12 with the protocol extended to cover all 84 fragments analysed here. CSCE peaks were analysed manually using Genotyper software to look for alterations in peak morphology compared with normal control samples. Batches of 27 patient samples were analysed with four normal controls for comparison and a negative control. Any fragments showing altered morphology during CSCE analysis or size changes during QF-PCR analysis were re-amplified individually from the patient DNA and sequenced using Big Dye terminator v3.1 chemistry (Applied Biosystems) to determine the nature of the sequence change. Sequence data were analysed using Mutation Surveyor software v2.61 (Softgenetics, State College, PA, USA). The entire process is summarized in Figure 1.

Flow chart summarizing the full mutation screening process.
Results
Assessment of multiplexes
Little optimization was needed for the dosage assay, as this technique was already established in the laboratory12 and was simply extended to cover the entire DMD coding region. Testing of the multiplexes was performed using normal control DNA samples and all gave dosage results that were consistent with all exons being present in their normal copy number at 22 cycles. An example multiplex is shown in Figure 2. All amplicons were found to give consistent dosage measurements, except for exon 63, therefore, a second set of exon 63 primers was included in the assay to overcome this problem. Attempts were made to establish one PCR cycle number, which could be used for both quantitative analysis and heteroduplex detection, using the concentration of DNA, which had previously been optimized for QF-PCR.12 However, it was found that increased cycle numbers above 26 showed dosage results with greater deviation from the expected results (data not shown), whereas heteroduplex detection efficiency was highest at 30 cycles.

An example multiplex (one of 12) containing 7 exons of the DMD gene. The peak area of each exon for all samples is exported in a table format from Genotyper software (Applied Biosystems) to a Microsoft Excel spreadsheet, where the dosage quotients for each exon are calculated relative to the dosage quotient of normal control samples.
Determination of FM-CSCE conditions
The 12 multiplexes were then used to determine the optimum FM-CSCE conditions on the ABI 3100. After testing various different polymers, temperatures, run voltages and capillary lengths (not described here), the optimum conditions were found to be those described in the methods section. Visual inspection of the peaks was carried out manually using Genotyper software (Applied Biosystems), comparing the morphology of each peak to that of the normal control peak for each exon. Testing on a panel of 31 different substitution mutations in 48 different male and female patients showed a pattern clearly different from the normal control in 47/48 samples (98%). One mutation could be detected in a male patient but the shift was more subtle in his mother's DNA sample and this was therefore regarded as a failure to detect the sequence change. This subtle difference illustrates the subjective nature of the approach. Figure 3 demonstrates some of the different peak patterns produced by different sequence changes in the same fragment, containing exon 14. Clearly, it is not possible to predict either the position or type of mutation from the heteroduplex pattern produced. The possibility that the presence of a polymorphism in a fragment may mask the presence of a mutation was considered. For example, there is a polymorphism in exon 21 of the DMD gene (c.2645G>A; [p.Gly882Asp]; allele frequency listed as 0.15 on the Leiden muscular dystrophy website www.dmd.nl). Figure 4 shows different combinations of mutation and polymorphism and demonstrates that this polymorphism has not masked the presence of these mutations.

CSCE traces of exon 14 in four samples with sequence changes and a normal control (sequence changes were determined in each case by sequencing of genomic DNA). Trace (a) (c.1684dupC; p.Gln562fs), is due to a one base pair insertion. Traces (b) (c.1615C>T; p.Arg539X) and (c) (c.1702C>T; p.Gln568X) are due to nonsense mutations. Trace (d) is due to two common polymorphisms in exon 14 and intron 14 (c.[1635A>G; 1704+51c>t]). Trace (e) is that of a normal control sample without either of the common polymorphisms.

CSCE traces of exon 21 in four samples with sequence changes listed (sequence changes were determined in each case by sequencing of genomic DNA). This exon contains a frequent variant c.2645G>A (p.Gly882Asp). Trace (a) shows a sample with a mutation (c.2797C>T; p.Gln933X) and the G allele; trace (b) shows a sample with a nonsense mutation (c.2650C>T; p.Gln884X) and the A allele; trace (c) shows a sample with no mutation and the A allele; trace (d) shows a sample with no mutation and the G allele. These can all be seen to produce different CSCE patterns, indicating that the presence of the polymorphism has not masked these mutations. This fragment (and many others) always consists of two peaks under non-denaturing conditions. This is thought to be due to the chemistry of the fluorescent dyes used.
Blind trial
Once the optimum conditions for CSCE had been established, a blind trial was carried out by applying the full mutation screen to 50 male and 50 female DNA samples, which had previously been characterized using alternative methods. The mutations and results obtained are shown in full in Supplementary Table 1 (Supplementary Information) and summarized in Table 1. All 84 previously known mutations were detected. In two cases (F15 and F19), females who had previously been shown to have duplications using a limited screen of 28 exons were actually discovered to have non-contiguous duplications when all 79 exons were screened. These results have been confirmed using MLPA. One sample (M27), which had previously only been screened for deletions and duplications, was found to have a nonsense mutation in exon 57 (this result was confirmed under diagnostic conditions). In 18 cases, the end points of a deletion or duplication were defined by testing all 79 exons rather than the sub-set of 28 exons previously tested.
Testing patient samples
The results are summarized in Table 2. The details of mutations detected can be found in the Supplementary Information (Supplementary Table 2) and have been deposited on the Leiden muscular dystrophy database (www.dmd.nl).
Pathogenic mutations were detected in 45 samples from a cohort of 58 males originally referred as DMD patients. Of the remaining 13 patients, two were shown subsequently to have large genomic inversions using the protein truncation test (unpublished data, manuscript in preparation); seven have since been assigned alternative clinical diagnoses of Limb Girdle Muscular Dystrophy type 2I (3), X-linked Emery–Dreifuss muscular dystrophy (1), Hereditary Motor and Sensory Neuropathy (1), Merosin-deficient congenital muscular dystrophy (1), Limb girdle muscular dystrophy (type unknown) (1); three have uncertain diagnoses (no muscle biopsy or creatine kinase measurements available); and one has a confirmed diagnosis (absent dystrophin staining on muscle) but the protein truncation test has not been carried out as no further biopsy material was available.
In 16 out of 51 samples from BMD patients, a pathogenic mutation was detected. Of the remaining 35 patients, 16 have now had alternative clinical diagnoses of Limb Girdle Muscular Dystrophy type 2I (8), a metabolic disorder of unknown type (1), Limb girdle muscular dystrophy (type unknown) (1), Emery–Dreifuss muscular dystrophy (2), raised creatine kinase during a transient episode of myoglobinuria (1) normal dystrophin staining (final clinical diagnosis unknown) (3); 12 have an uncertain diagnosis and we are unable to follow these patients up any further, and seven have an almost certain diagnosis of BMD based on muscle biopsy results or raised creatine kinase levels. Unfortunately, no muscle biopsy is available from any of these individuals to carry out further testing.
The mutation detection rates based on this testing are 78% (45 out of 58) for individuals referred initially with a diagnosis of DMD, rising to 94% (45 out of 48) where this diagnosis has been confirmed. For individuals referred with an initial diagnosis of BMD, the detection rate is 31% (16 out of 51), rising to 70% (16 out of 23) where the diagnosis has been confirmed (Table 2).
Discussion
We have developed and applied a DNA-based mutation screening technique applicable for high-throughput mutation screening of any gene. It is a rapid simultaneous screen for detecting nearly all mutation types, which can be tailored to any number of exons. The CSCE method allows samples to be run under only one set of electrophoretic conditions to detect all mutations, unlike other techniques such as dHPLC or DOVAM, which often require a set of samples to be run under a number of different conditions to maximize mutation detection. In our experience, a full mutation screen of the dystrophin gene using this technique, including sequencing of possible variants, can be completed for urgent cases in 10 working days.21 However, this is an inefficient use of time and reagents.
We have also successfully applied this mutation screening technique to exon 11 of the breast cancer susceptibility gene, BRCA1, and to the X-linked Alport syndrome gene, COL4A5. In the case of COL4A5, tagged primers were used to reduce costs as this removes the need for fluorescently labelled primers for each fragment to be analysed.
Since this project began, MLPA kits have become commercially available to detect whole exon deletions and duplications for various genes, including DMD.14 MLPA can be used to confirm any single exon deletions and duplications detected by QF-PCR. An alternative approach would be to screen for large deletions and duplications using MLPA, use QF-PCR to confirm the presence of single exon deletions and then to use CSCE to screen for point mutations if no mutation was identified using MLPA. However, this would involve multiple testing and running of samples compared with the approach described here.
The number of fragments requiring sequencing following the CSCE analysis depends on whether a pathogenic mutation is identifiable or not, since fragments harbouring mutations generally stand out with different electrophoretic traces compared with controls. Taking the data from a standard batch of 27 samples, an average of four exons were sequenced (5% of fragments) where pathogenic mutations were identified, whereas where no mutation was identified this number rose to an average of 21 fragments (26% of fragments).
The predicted sensitivity of this mutation screening approach is comparable with the prospective data obtained from screening 109 patients, who had previously been screened for deletions and duplications of 28 exons in the hotspot regions. Mutations were identified in 61 out of 109 patients screened using this technique (see Table 2 for summary and Supplementary Table 2 Supplementary Information for full mutation details).
If we concentrate on patients with no whole exon deletions or duplications, the CSCE approach detected small mutations in 38/51 (75%) of patients originally referred with DMD and only 13/48 (27%) of those referred with BMD. However, if we ignore the 23 patients with a non-Xp21 muscular dystrophy and the 15 individuals in whom the diagnosis was not clinically confirmed, then extrapolate assuming that whole exon deletions/duplications are present in a minimum of 70% of DMD patients and 80% of BMD patients, combining a full deletion/duplication screen with CSCE will detect a mutation in 98% of DMD patients and 93% of BMD patients.
Our overall results show that the mutation detection rate is lower for individuals referred with BMD rather than DMD, both in cases where a clinical diagnosis had been confirmed with a muscle biopsy and in non-confirmed cases. This difference may be because of poor diagnosis, or perhaps a greater proportion of mutations causing BMD are of a type which cannot be detected using this technique: that is inversions, deeply intronic mutations, those involving the promoter, or mutations in other genes which produce proteins that interact with dystrophin.
A distinct advantage of this method is that it does not require a muscle biopsy to be taken for the majority of clinically confirmed cases where a mutation can be detected using DNA. In addition, screening can be offered to the 20% of families where there are no samples available from an affected male, by testing obligate carriers or females most likely to be carriers. Screening of obligate carriers or mothers and sisters of males who are isolated cases can either identify a mutation or greatly reduce carrier risks. This approach has been successful for a number of families where carrier risks were ambiguous before screening and has enabled prenatal diagnosis or reassurance that relatives are not carriers of a DMD mutation21 (and unpublished cases).
In addition to genetic counselling, knowledge of the exact mutation in a particular individual will be essential for some of the gene therapies currently being developed. For example, exon skipping therapies22, 23 require the exact end points of a large deletion or duplication to be defined, or to know which exon a point mutation is in to be able to therapeutically remove the affected region of a gene from the transcript. The technique described here makes that possible for the DMD gene and any other large genes it is applied to, where previously this may not have been the case.
This technique is applicable to any gene, particularly large genes with heterogeneous mutation spectrums. It significantly decreases the time taken for a mutation screen of a large gene for unknown mutations, due to the combination of both techniques with one PCR product, multiplexing of fragments and use of a high throughput genetic analyser. It can be applied to screen more than one gene simultaneously where this is appropriate for the clinical diagnosis, for example the BRCA1 and BRCA2 genes, and will enable laboratories to reduce the time taken to perform screening of large genes for all mutation types.
References
Lu QL, Mann CJ, Lou F et al: Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat Med 2003; 9: 1009–1014.
Welch EM, Barton ER, Zhuo J et al: PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007; 447: 87–91.
Abbs S, Bobrow M : Analysis of quantitative PCR for the diagnosis of deletion and duplication carriers in the dystrophin gene. J Med Genet 1992; 29: 191–196.
Van Essen AJ, Kneppers AL, van der Hout AH et al: The clinical and molecular genetic approach to Duchenne and Becker muscular dystrophy: an updated protocol. J Med Genet 1997; 34: 805–812.
Worton RG, Thompson MW : Genetics of Duchenne muscular dystrophy. Annu Rev Genet 1988; 22: 601–629.
Bushby KM, Thambyayah M, Gardner-Medwin D : Prevalence and incidence of Becker muscular dystrophy. Lancet 1991; 337: 1022–1024.
Aartsma-Rus A, Van Deutekom JCT, Fokkema IF, Van Ommen G-JB, den Dunnen JT : Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006; 34: 135–144.
Monaco AP, Bertelson CJ, Middlesworth W et al: Detection of deletions spanning the Duchenne muscular dystrophy locus using a tightly linked DNA segment. Nature 1985; 316: 646–650.
Chamberlain JS, Gibbs Ra, Ranier JE, Nguyen PN, Caskey CT : Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res 1988; 16: 11141–11156.
Beggs AH, Koenig M, Boyce FM, Kunkel LM : Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet 1990; 86: 45–48.
Abbs S, Yau SC, Clark S, Mathew CG, Bobrow M : A convenient multiplex PCR system for the detection of dystrophin gene deletions: a comparative analysis with cDNA hybridisation shows mistypings by both methods. J Med Genet 1991; 28 : 304–311.
Yau SC, Bobrow M, Mathew CG, Abbs SJ : Accurate diagnosis of carriers of deletions and duplications in Duchenne/Becker muscular dystrophy by fluorescent dosage analysis. J Med Genet 1996; 33: 550–558.
White S, Kalf M, Liu Q et al: Comprehensive detection of genomic deletions and duplications in the DMD gene, by use of multiplex amplifiable probe hybridization. Am J Hum Genet 2002; 71: 365–374.
Shouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G : Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002; 30: e57.
Roest PAM, Roberts RG, Sugino S, van Ommen GJB, den Dunnen JT : Protein truncation test (PTT) for rapid detection of translation-terminating mutations. Hum Mol Genet 1993; 2: 1719–1721.
Bennett RR, den Dunnen J, O'Brien KF, Darras BT, Kunkel LM : Identification of mutations in the dystrophin gene via automated DHPLC screening and direct sequencing. BMC Genet 2001; 2: 17.
Mendell JR, Buzin CH, Geng J et al: Diagnosis of Duchenne dystrophy by enhanced detection of small mutations. Neurology 2001; 57: 645–650.
Flanigan KM, von Niederhausern A, Dunn DM, Alder J, Mendell JR, Weiss RB : Rapid direct sequence analysis of the Dystrophin gene. Am J Hum Genet 2003; 72: 931–939.
Hofstra RMW, Mulder IM, Vossen R et al: DGGE-based whole-gene mutation scanning of the dystrophin gene in Duchenne and Becker Muscular Dystrophy patients. Hum Mutat 2004; 23: 57–99.
Hamed SA, Hoffman E : Automated sequence screening of the entire dystrophin cDNA in Duchenne dystrophy: point mutation detection. Am J Med Genet 2006; 141B: 44–50.
Ashton E, Deans Z, Yau SC, Abbs S : A novel and rapid mutation screening approach facilitates prenatal diagnosis. Prenat Diagn 2005; 25: 425–426.
Blankinship MJ, Gregorevic P, Chamberlain JS : Gene therapy strategies for Duchenne muscular dystrophy utilizing recombinant adeno-associated virus vectors. Mol Ther 2006; 13: 241–249.
McClorey G, Fletcher S, Wilton S : Splicing intervention for Duchenne muscular dystrophy. Curr Opin Pharmacol 2005; 5: 1–6.
Acknowledgements
We thank all genetics centres who referred patients to us for testing, Dr Roli Roberts for his helpful advice and Professor Francesco Muntoni for his continued collaboration. This work was funded by the Guy's and St Thomas' charity.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Ashton, E., Yau, S., Deans, Z. et al. Simultaneous mutation scanning for gross deletions, duplications and point mutations in the DMD gene. Eur J Hum Genet 16, 53–61 (2008). https://doi.org/10.1038/sj.ejhg.5201916
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201916
Keywords
This article is cited by
-
Duchenne muscular dystrophy caused by a frame-shift mutation in the acceptor splice site of intron 26
BMC Medical Genetics (2016)
-
Correlations between long inverted repeat (LIR) features, deletion size and distance from breakpoint in human gross gene deletions
Scientific Reports (2015)
-
Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center
Journal of Human Genetics (2010)
-
A novel custom high density-comparative genomic hybridization array detects common rearrangements as well as deep intronic mutations in dystrophinopathies
BMC Genomics (2008)
