
Abstract
Background
There is now greater involvement of children in drug trials to ensure that paediatric medicines are supported by sound scientific evidence. The safety of the participating children is of paramount importance. Previous research shows that these children can suffer moderate and severe adverse drug reactions (ADRs) in clinical trials, yet very few of the trials designated a data safety monitoring board (DSMB) to oversee the trial.
Methods
Safety data from a systematic review of paediatric drug randomised controlled trials (RCTs) published in 2007 were analysed. All reported adverse events (AEs) were classified and assessed to determine whether an ADR had been experienced. ADRs were then categorised according to severity. Each trial report was examined as to whether an independent DSMB was in place.
Results
Of the 582 paediatric drug RCTs analysed, 210 (36%) reported that a serious AE had occurred, and in 15% mortality was reported. ADRs were detected in more than half of the RCTs (305); 66 (11%) were severe, and 79 (14%) were moderate. Severe ADRs involved a wide range of organ systems and were frequently associated with cytotoxic drugs, antiparasitics, anticonvulsants and psychotropic drugs. Two RCTs reported significantly higher mortality rates in the treatment group. Only 69 (12%) of the RCTs stated there was a DSMB. DSMBs terminated five RCTs and changed the protocol in one.
Conclusions
Children participating in drug RCTs experience a significant amount and a wide range of ADRs. DSMBs are needed to ensure the safety of paediatric participants in clinical drug trials.
Similar content being viewed by others

Introduction
Recent changes in US and European drug regulation have provided a stimulatory environment for paediatric clinical drug trials to be performed [1, 2]. As a result, larger numbers of paediatric clinical trials have been conducted providing valuable information for the judicious use of medicines in children [3–5]. However, when participating in clinical trials, paediatric patients are exposed to a risk of experiencing adverse drug reactions (ADRs). This is a primary concern to parents, clinicians and regulatory agencies due to the vulnerable nature of the paediatric population and that participation of children in these trials are via proxy consent of their caregivers. However, surprisingly little work has attempted to shed light on the toxicity experienced of paediatric participants in drug trials. A review of >700 paediatric drug trials published between 1996 and 2002 found that ADRs occurred in more than a third of the trials. Although 11% of the trials reported that moderate or severe ADRs occurred, only 2% of the trials mentioned that a data safety monitoring board (DSMB) was formed to oversee the safety of the study [6]. This previous study, however, was not a comprehensive systematic review in that only one database (Medline) was used and trials in the areas of oncology and HIV were excluded. We therefore felt it was appropriate to perform a comprehensive systematic review of drug toxicity within paediatric drug randomised controlled trials (RCTs) published in 2007 [7].
Methods
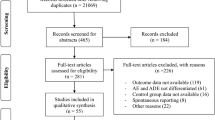
The method used to establish the database of paediatric drug RCTs has been previously described [7]. Briefly, Medline, Embase and Cochrane Collaboration Central Register databases were searched electronically to systematically identify RCTs published in 2007. Validated search strategies derived from those developed by the Cochrane Collaboration [8] and the Hedges Team [9] were used. The search was limited to drug trials and involving paediatric patients. The resulting citations were hand searched, and full-text papers were obtained for all studies deemed relevant. Supplementary material was retrieved whenever available. Each RCT report was carefully read to evaluate whether safety monitoring was mentioned in the Methods section. Any mention of the words safety, adverse effect/event/experience/reaction, side-/unwanted effect, toxicity or any indication that adverse events were monitored was noted. Whether any adverse events (AEs) were detected was determined from carefully reading the Results section or supplementary information when available. The definition and classification of AEs used in this study were based on guidelines produced by the European Agency for the Evaluation of Medicinal Products (EMA) and the International Conference for Harmonisation (ICH) [6]. An AE was defined as any untoward medical occurrence in a patient or clinical investigation participant administered a pharmaceutical product and did not necessarily need to have a causal relationship with this treatment. AEs are classified as serious, significant or mild according to the following groupings [6]:
-
1.
Serious AE (SAE): any untoward medical occurrences at any dose that results in death, is life-threatening, requires in-patient hospitalisation or prolongation of existing hospitalisation, results in persistent or significant disability/incapacity, or is a congenital anomaly/birth defect.
-
2.
Significant AE: haematological and other laboratory abnormalities and any AE that led to an intervention, including withdrawal of drug treatment, dose reduction or significant additional concomitant therapy.
-
3.
Mild AE: any AE occurring that did not need any intervention.
The number of papers reporting any mortality was noted. Any trial that was discontinued was also noted and the reason for discontinuation determined. The most serious AE in each report was determined and used to stratify the RCTs. All trials reporting a serious AE were further reviewed by two paediatric clinical pharmacologists (IC and HMS) independently to judge whether any of the serious AEs were possible ADRs. The decision by each reviewer was noted, and when there was disagreement, a consensus was obtained from joint discussions. An ADR was defined as an AE thought to be linked in either time or dose to a drug given to that patient. Each RCT included in this study was assessed as to whether a possible ADR had occurred and were classified according to the highest severity of ADR in the report. The classification used for ADRs was as follows [6, 10]:
-
1.
Severe: fatal or potentially life threatening or causing permanent disability.
-
2.
Moderate: requiring treatment or prolonging stay in hospital.
-
3.
Mild: no treatment required and no effect on length of stay in hospital.
In RCTs where ADRs were detected, the following characteristics were analysed:
-
1.
The World Health Organisation (WHO) Anatomical–Therapeutic–Chemical (ATC) classification category.
-
2.
Population age group being studied.
-
3.
Sample size of the study.
All RCT reports were checked to determine whether a DSMB or an independent safety evaluator was involved in overseeing the trial. In addition, it was determined whether any interim analysis [11] was performed or whether a stopping rule [12] for the trials was designated. These were assumed to be present whenever a DSMB was mentioned. Data were analysed and stored using SPSS version 16.0 (SPSS Inc., Chicago, IL, USA). Categorical variables are described with frequencies or percentages. Fisher’s exact test was used to calculate p values for differences in proportions.
Results
Safety data from 582 paediatric RCTs were analysed. Epidemiological and methodological characteristics of these trials have been previously described [7]. Five hundred and forty trials involved infants and children, and 42 (7%) involved neonates. Approximately two thirds of the trials (375, 64%) mentioned safety monitoring in the Methods section. The majority of the trials (463, 80%) reported on AEs including studies stating that no AEs had occurred. Over one third of the trials (210, 36%) reported that an SAE had occurred in the trial. Eighty-seven RCTs (15%) reported that mortality had occurred during the trial. Twenty-two RCTs involving cytotoxic drugs (56% of the total number of oncology RCTs) reported mortalities. Six of the 13 trials involving cardiovascular drugs (46%) reported mortalities.
ADRs were considered to have occurred in more than half of the trials (305, 52%), whereas the remainder were either determined to have experienced no ADRs (141, 24%) or were found to be impossible to judge whether an ADR had occurred (136, 23%). In just over a quarter of the trials (160, 28%), only mild ADRs occurred. Moderate ADRs occurred in 79 (14%) RCTs. Severe ADRs were found in 66/582 (11%) of RCTs involving children. Severe ADRs occurred most frequently in oncology RCTs (49%, 19/39 trials) and cardiovascular drug RCTs (15%, 2/13 trials) (Table 1).
The severe ADRs detected included most of the major organ systems and a wide range of medicines. The severe ADRs detected and the drugs involved are shown in Table 2 (infants and children) and Table 3 (neonatal). There was no significant difference between the proportions of neonatal and nonneonatal RCTs in which severe ADRs were detected: 7/42 (17%) vs 59/540 (11%, p = 0.31).
Only 69 (12%) RCTs documented that a DSMB or independent safety evaluator was involved. An additional four trials mentioned either termination rules or that interim analysis was done but without specifically mentioning the presence of a DSMB. Significantly more trials that mentioned an SAE occurring documented that a DSMB was present in comparison to trials with no SAEs (55/210 vs 14/372 trials, p < 0.05). DSMBs terminated five RCTs and changed the protocol of one after an episode of toxicity [13]. Three RCTs were terminated due to the risk of toxicity [14–16], one was terminated for lack of efficacy [17] and another for administrative reasons [18] (Table 4). Four of these trials involved neonates.
Two RCTs reported significantly higher mortality rates in the treatment group. One of these involved chemotherapy for germ-cell tumours and was terminated by the DSMB [15]. The other was a trial comparing magnesium sulphate infusions to placebo in 499 patients 14 years of age or older who were admitted to a trauma centre with traumatic brain injury [19]. The trial involving magnesium sulphate did not appear to have a DSMB, and it is difficult to establish the number of paediatric patients who died within this trial.
Discussion
The safety of medicines given to children has received significant attention in recent years [20]. The safe and effective use of paediatric medications requires adequate information gained from clinical trials involving children themselves [21]. When this fact was highlighted by health professionals, governments were prompted to take regulatory action and provide incentives towards the drug industry who are the main sponsors of drug research [22]. This has led to large numbers of children participating in drug studies, including RCTs [7]. In RCTs in which children are involved, the potential of harm caused by ADRs is a primary concern for parents, investigators, regulators and sponsors. ADRs affect a significant number of patients being treated with drugs. A large meta-analysis found that 10.9% of all hospitalised patients (both adult and paediatric) experience a severe ADR, and fatal ADRs were between the fourth and sixth leading cause of death [23].
This review demonstrates that ADRs occurred in more than half (52%) of drug RCTs involving paediatric patients that were published in 2007. It is important to note that this percentage does not indicate the risk of ADRs for children taking part in RCTs but merely the proportion of trials in which ADRs were detected [6]. Previous studies have shown that the overall incidence of ADRs in the paediatric population is 634 per million children per year [24] and approximately 10% for hospitalised children [25]. It is important that ADRs are assessed in the setting of a trial so they can be established before the medicine is used in clinical practice.
Significant drug toxicity (moderate or severe ADRs) was detected in a quarter (25%) of the trials in our study. This is higher than previously reported (11%) [6]. This study is the first systematic review published of safety in paediatric clinical trials. Previous reviews have not been systematic in that they have either not included all paediatric RCTs or not used more than one database [6, 26] to capture all the available evidence. In our study, antineoplastic drug trials accounted for roughly one third (19/66) of the RCTs in which severe ADRs were detected. This is not surprising, as cytotoxic drugs—because of their mode of action—are invariably associated with significant drug toxicity.
Clinical trials of cytotoxic drugs are essential. It is important, however, that parents and children are aware of the risk for drug toxicity within a clinical trial and/or a treatment regime. This can then be weighed against the child’s clinical condition. In a large number of RCTs in this study, it was impossible to ascertain whether an ADR had occurred from the safety data reported. Twenty percent of the RCTs in our database did not report any safety data. The inadequate reporting of safety data from RCTs in both adults and children has been noted previously [26–28]. RCTs are usually not powered to detect ADRs [29]. Despite this, it is important that RCTs adequately and transparently report safety data. This then allows for meta analysis later when suitable and gives clinicians appropriate evidence for their prescribing decisions.
There is persisting concern that very few paediatric RCTs in our review documented that DSMBs were formed to oversee the trial. The studies in which DSMBs intervened clearly demonstrated the vital role they play in ensuring the safety of the participating patients. The latest guidelines on ethical conduct of paediatric clinical trials now state that DSMBs should be created for all phase 3 clinical trials involving paediatric patients [30]. Of the 582 RCTs published in 2007, five were terminated by a DSMB. Unfortunately, not all trials are published and, where a trial has been terminated or there is significant toxicity, authors are probably less likely to publish their findings [31].
Therefore, safety issues must be a key consideration at the design stage of a clinical trial. Stopping rules need to be determined at the outset, with a termination plan in place in case of severe ADRs occurring [32]. An independent and effective DSMB should be instituted. There are clear guidelines available as to who should be members of a DSMB and their roles [33].
Three out of the five terminated trials were neonatal RCTs. Although neonatal RCTs constituted only 7% of the trials in our database [7], severe ADRs were prominent in the neonatal RCTs reviewed. It is recognised that the physiology of the neonate is very different from that of older children and may predispose to certain drug toxicities [34]; however, more research is needed to elucidate whether neonates are more at risk of ADRs than are older children.
In conclusion, findings from this study confirmed that moderate and severe ADRs occur in 25% of RCTs involving children. Unfortunately, very few of these RCTs document the presence of a DSMB. All clinical trials involving the paediatric population should have a designated DSMB. The role of the DSMB is crucial in protecting children participating in RCTs, especially in the current situation where there is a growing effort to formally evaluate drugs in children to provide them with safe and effective medicines.
References
Smyth RL (2007) Making a difference: the clinical research programme for children. Arch Dis Child 92(10):835–837
Hoppu K (2008) Paediatric clinical pharmacology - at the beginning of a new era. European Journal of Clinical Pharmacology 64:201–205
Roberts R, Rodriguez W, Murphy D, Crescenzi T et al (2003) Pediatric drug labeling: improving the safety and efficacy of pediatric therapies. JAMA 290(7):905–911
Rodriguez W, Selen A, Avant D, Chaurasia C, Crescenzi T, Gieser G et al (2008) Improving Pediatric Dosing Through Pediatric Initiatives: What We Have Learned. Pediatrics 121(3):530–539
Sammons HM, Choonara I (2005) Clinical trials of medication in children, 1996–2002. European Journal of Clinical Pharmacology 61(2):165–167
Sammons HM, Gray C, Hudson H, Cherrill J, Choonara I et al (2008) Safety in paediatric clinical trials–a 7-year review. Acta Paediatrica 97(4):474–477
Nor Aripin KNB, Choonara I, Sammons HM (2010) A systematic review of paediatric randomised drug trials published in 2007. Archives of Diseases in Childhood 95(6):469–473
The Cochrane Collaboration. Cochrane Handbook for Systematic Reviews of Interventions John Wiley & Sons, Ltd, 2008.
Wong SS, Wilczynski NL, Haynes RB, Wong SSL, Wilczynski NL, Haynes RB (2006) Comparison of top-performing search strategies for detecting clinically sound treatment studies and systematic reviews in MEDLINE and EMBASE. Journal of the Medical Library Association 94(4):451–455
Turner S, Nunn AJ, Fielding K, Choonara I (1999) Adverse drug reactions to unlicensed and off-label drugs on paediatric wards: a prospective study. Acta Paediatrica 88:965–968
Fossa SD, Skovlund E (2000) Interim analyses in clinical trials: why do we plan them? Journal of Clinical Oncology 18:4007–4008
Hedenmalm K, Melander H, Alvan G (2008) The conscientious judgement of a DSMB–statistical stopping rules re-examined. European Journal of Clinical Pharmacology 64:69–72
Lands LC, Milner R, Cantin AM, Manson D, Corey M (2007) High-dose ibuprofen in cystic fibrosis: Canadian safety and effectiveness trial. Journal of Pediatrics 151:249–254
Van Meurs KP, Hintz SR, Ehrenkranz RA, Lemons JA, Ball MB, Poole WK et al (2007) Inhaled nitric oxide in infants >1500 g and <34 weeks gestation with severe respiratory failure. Journal of Perinatology 27(6):347–352
Lorch A, Kollmannsberger C, Hartmann JT, Metzner B, Schmidt-Wolf IG, Berdel WE et al (2007) Single versus sequential high-dose chemotherapy in patients with relapsed or refractory germ cell tumors: a prospective randomized multicenter trial of the German Testicular Cancer Study Group. Journal of Clinical Oncology 25(19):2778–2784
Bonsante F, Latorre G, Iacobelli S, Forziati V, Laforgia N, Esposito L et al (2007) Early low-dose hydrocortisone in very preterm infants: A randomized, placebo-controlled trial. Neonatology 91:217–221
Nadel S, Goldstein B, Williams MD, Dalton H et al (2007) Drotrecogin alfa (activated) in children with severe sepsis: a multicentre phase III randomised controlled trial. Lancet 369:836–843
Sullivan PB, Lewindon PJ, Cheng C, Lenehan PF, Kuo BS, Haskins JR et al (2007) Intestinal mucosa remodeling by recombinant human epidermal growth factor(1–48) in neonates with severe necrotizing enterocolitis. Journal of Pediatric Surgery 42(3):462–469
Temkin NR, Anderson GD, Winn RH, Ellenbogen RG, Britz GW, Schuster J, Lucas T, Newell DW, Mansfield PN, Machamer JE, Barber J, Dikmen SS (2007) Magnesium sulfate for neuroprotection after traumatic brain injury: a randomised controlled trial. Lancet Neurology 6(1):29–38
World Health Organisation. Promoting safety of medicines for children. Available at: http://www.who.int/medicines/…/essentialmedicines/Promotion_safe_med_childrens.pdf. (Accessed November, 2009).
Budetti P (2003) Ensuring safe and effective medications for children. JAMA 290(7):950–951
Choonara I (2007) Regulation of drugs for children in Europe. BMJ 335:1221–1222
Lazarou J, Pomeranz BH, Corey PN (1998) Incidence of adverse drug reactions in hospitalized patients: A Meta-analysis of prospective studies. JAMA 279:1200–1205
Bárzaga Arencibia Z, Novoa Sotomayer D, Caballero Mollinedo, Choonara I, Fernández Manzano E, López Leyva A (2010) Adverse drug reactions in children in Camagüey Province, Cuba. Arch Dis Child 95:474–477
Clavenna A, Bonati M (2009) Adverse drug reactions in childhood: a review of prospective studies and safety alerts. Arch Dis Child 94:724–728
de Vries, T.W., van Roon, E.N. Low quality of reporting adverse drug reactions in paediatric randomised controlled trials. Arch Dis Child 2010, Published online first 15 June 2010. Available at: http://adc.bmj.com/content/early/2010/06/14/adc.2009.175562.short?rss=1. (Accessed August 2010).
Ioannidis JPA (2009) Adverse events in randomized trials: neglected, restricted, distorted, silenced. Arch Intern Med 169:1737–1739
Anderson M, Choonara I (2010) A systematic review of safety monitoring and drug toxicity in published randomised controlled trials of antiepileptic drugs in children over a 10-year period. Arch Dis Child 95:731–738
Tsang R, Colley L, Lynd LD (2009) Inadequate statistical power to detect clinically significant differences in adverse event rates in randomized controlled trials. J Clin Epidemiol 62:609–616
World Health Organization. Operational guidelines for the establishment and functioning of data and safety monitoring boards. Available at: http://apps.who.int/tdr/svc/publications/tdr-research-publications/operational-guidelines. (Accessed October, 2009).
Cousins D, Clarkson A, Conroy S, Choonara I (2002) Medication errors in children – an eight year review using press reports. Paed Perinat Drug Ther 5:52–58
Grant A (2004) Stopping clinical trials early BMJ 329:525
DAMOCLES Study Group (2005) A proposed charter for clinical trial data monitoring committees: helping them to do their job well. Lancet 365:711–722
McIntyre J, Choonara I (2004) Drug toxicity in the neonate. Biol Neonate 86:218–221
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Nor Aripin, K.N.B., Choonara, I. & Sammons, H.M. Systematic review of safety in paediatric drug trials published in 2007. Eur J Clin Pharmacol 68, 189–194 (2012). https://doi.org/10.1007/s00228-011-1112-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-011-1112-6