Article Text

Abstract
Objective This study aimed to investigate the prevalence and clinical characteristics of monogenic disease in paediatric patients with a predominant respiratory phenotype.
Methods Exome sequencing was performed in a cohort of 971 children with a predominant respiratory phenotype and suspected genetic aetiology. A total of 140 positive cases were divided into subgroups based on recruitment age and the primary biological system(s) involved.
Results There were 140 (14.4%) patients with a positive molecular diagnosis, and their primary clinical manifestations were respiratory distress (12.9%, 18 of 140), respiratory failure (12.9%, 18 of 140) and recurrent/persistent lower respiratory infections (66.4%, 93 of 140). Primary immunodeficiency (49.3%), multisystem malformations/syndromes (17.9%), and genetic lung disease (16.4%) were the three most common genetic causes in the cohort, and they varied among the age subgroups. A total of 72 (51.4%) patients had changes in medical management strategies after genetic diagnosis, and the rate in those with genetic lung disease (82.6%, 19 of 23) was far higher than that in patients with genetic disease with lung involvement (45.3%, 53 of 117) (p=0.001).
Conclusion Our findings demonstrate that exome sequencing is a valuable diagnostic tool for monogenic diseases in children with a predominant respiratory phenotype, and the genetic spectrum varies with age. Taken together, genetic diagnoses provide invaluable clinical and prognostic information that may also facilitate the development of precision medicine for paediatric patients.
- epidemiology
- genetics
- information technology
- neonatology
- therapeutics
Data availability statement
All data relevant to the study are included in the article or uploaded as supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
What is already known on this topic?
Continuous progress has been made towards our understanding of genetic lung disease and genetic disease with lung involvement, especially the former, with substantial efforts and resources devoted to the research.
The application of the next-generation sequencing in cohort studies in paediatric respirology is relatively rare, and little is known about its value for diagnosis and clinical management of paediatric patients with a predominant respiratory phenotype.
What this study adds?
Genetic disorder spectrum of monogenic disease in paediatric patients with a predominant respiratory phenotype varies with age.
Genetic diagnoses provide invaluable clinical and prognostic information that may also facilitate the development of precision medicine for paediatric patients.
Introduction
Respiratory diseases in children are the most common reason for hospitalisation. They are associated with high morbidity and mortality worldwide, and adversely impact childhood quality of life, education and physical activity. With the improvements in molecular diagnostic techniques, there has been an increased recognition of the importance of genetic testing in paediatric respiratory diseases. However, the current awareness of the issue is mainly restricted to genetic lung disease (GLD), including alveolar capillary dysplasia with misalignment of pulmonary veins, surfactant metabolism dysfunction (SMD), pulmonary alveolar proteinosis, hereditary haemorrhagic telangiectasia, cystic fibrosis (CF) and primary ciliary dyskinesia (PCD).1 An excellent example is CF, which has been the focus of long-standing research efforts that have led to significant progress in diagnosis and treatment during the past few decades.2
In addition, respiratory management of genetic disease with lung involvement (GDL) has received relatively little attention, especially the respiratory management after receiving molecular diagnosis, because the focus has been directed on primary disease in most cases. GDLs include system-related genetic disorders, such as primary immunodeficiency, neuromuscular diseases, inherited metabolic diseases (IMDs) and connective tissue diseases; among them, the early non-specific and overlapping respiratory phenotypes pose enormous challenge for physicians.
Therefore, a large-scale investigation on monogenic diseases in children with a predominant respiratory phenotype is needed to enhance the knowledge about clinical characteristics, genotype, management, and outcome of these diseases and improve patient care and prognosis. In the present study, we analysed 140 patients (14.4%) who received a molecular diagnosis in our cohort of 971 patients with suspected genetic causes of respiratory manifestations. The objectives of the study were to: (1) investigate the prevalence of monogenic diseases in children with a predominant respiratory phenotype; (2) reveal genotype–phenotype correlations and (3) determine the value of genetic diagnoses for the development of precision medicine for children.
Materials and methods
Participants
Patients who were undiagnosed following routine clinical procedure and were suspected to have genetic disorders between 1 January 2015 and 31 December 2019 in the Children’s Hospital of Fudan University were enrolled if they had one of the following respiratory phenotypes: (1) respiratory distress after birth; (2) dyspnoea, progressive respiratory failure and/or oxygen dependence; and (3) recurrent/persistent lower respiratory infections (RLRIs/PLRIs). A detailed study protocol is described in the online supplemental methods. Informed consent was obtained from the parents of patients who were recruited in the present study.
Supplemental material
Exome sequencing and data analysis
Details of sequencing and alignment processing are described in the online supplemental methods. The bioinformatics analysis pipeline that we adopted had been previously reported.3 Rare sequence variants (minor allele frequency ≤1%) affecting genes known to cause Mendelian disorders after the above screening were classified in accordance with the American College of Medical Genetics and Genomics guidelines.4
Statistical analysis
Differences in subject characteristics were determined using the Χ2 or Fisher’s exact test when comparing three age groups, and via Mann-Whitney U test when comparing two patient groups (SPSS V.26.0). The significance level was set at a p value of 0.05.
Results
Demographic and clinical characteristics of the patients
In the entire cohort of 971 patients, a total of 140 (14.4%) patients received a positive molecular diagnosis and were included for further analysis (online supplemental figure S1). There were 98 (70%) boys and 42 (30%) girls, with a mean age at enrolment of 3.6 years (range: 0 days–17.8 years). The main respiratory manifestations at inclusion were respiratory distress (n=18, 12.9%), respiratory failure (n=18, 12.9%), and RLRI/PLRI (n=93, 66.4%), with the mean onset age shown in table 1. Patients were grouped based on enrolment age, and basic information and prevalent phenotype in each group are shown in table 1 and online supplemental results.
Characteristics of 140 genetic-positive patients in the study among subgroups
Spectrum of genetic disorders
There were 173 pathogenic/likely pathogenic variants spanning 54 genes in the 140 patients diagnosed with genetic disorders. The proportion of the different inheritance modalities was seen in online supplemental table S1. The origins of variants in 103 of the diagnosed patients are listed in online supplemental table S2, whereas this information was unavailable for the remaining 37 diagnosed patients because the DNA samples from their parents were not obtained.
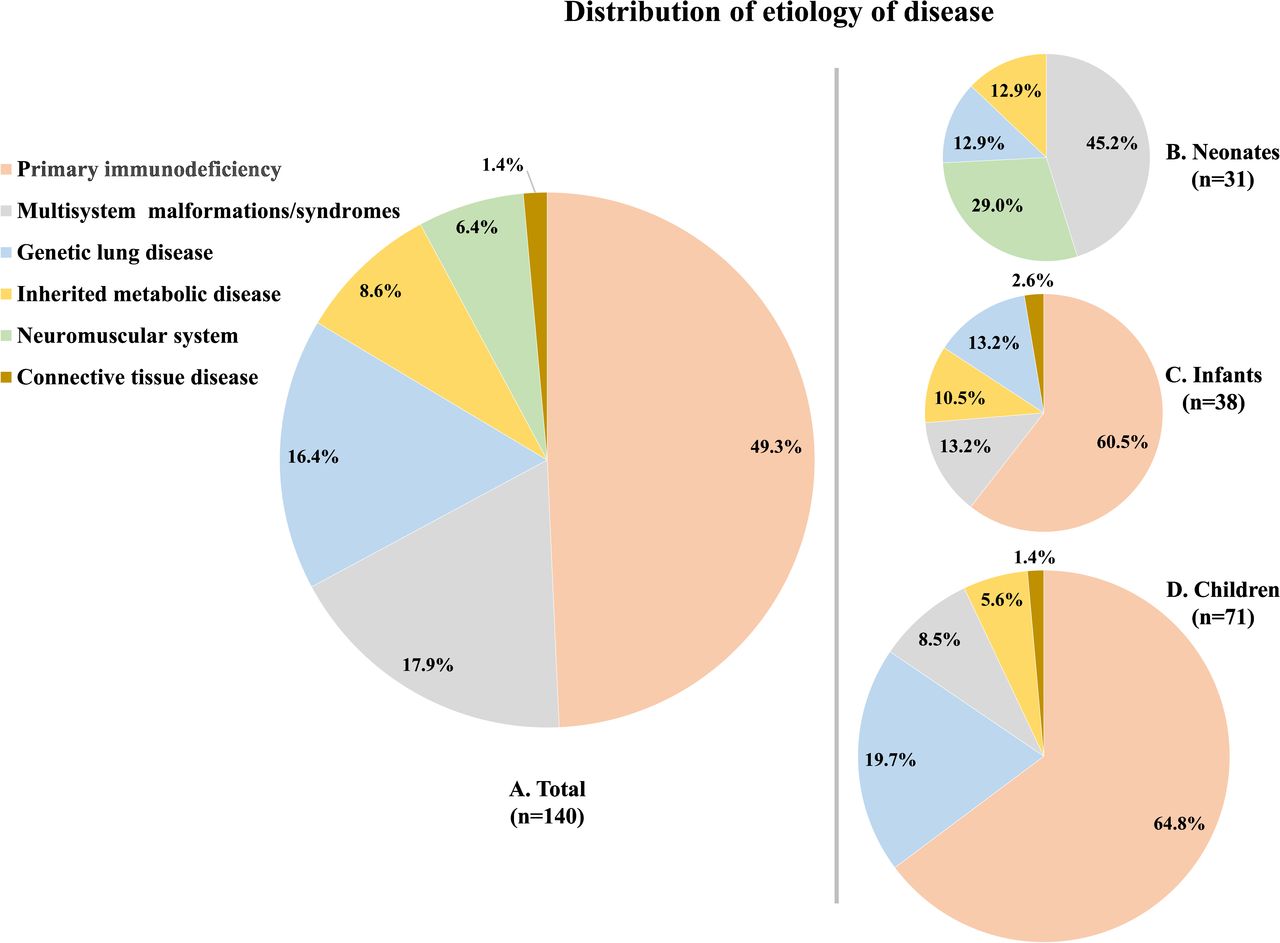
Genetic disorders were classified into six categories according to the primary system(s) involved in the disease aetiology, primarily determined by 54 phenotype-related genes that were diagnosed, as follows: GLD, primary immunodeficiency (PID), multisystem malformations/syndromes (MM/S), IMD, neuromuscular disease and connective tissue disease. The latter five were collectively referred to as GDL. GLD included 23 patients (16.4%), and GDL comprised 117 patients (83.6%). Comparisons of the clinical and demographic characteristics between the two groups are shown in table 2. PID (49.3%, 69 of 140), MM/S (17.9%, 25 of 140) and GLD (16.4%, 23 of 140) were identified as the three most common genetic causes. The main cause of age subgroups is seen in figure 1.

Distribution of disease spectrum among neonate, infant and children groups. (A) Distribution of disease spectrum of all 140 molecular-positive patients; (B) disease spectrum of neonate group; (C) disease spectrum of infant group; (D) disease spectrum of children group.
Comparison of clinical severity and medical management impact of molecular diagnosis between patients diagnosed with genetic lung disease and genetic disease with lung involvement
The distribution of the causative genes identified in the GLD group, GDL group and age grouping source is shown in figure 2. SMDs caused by SFTPC and ABCA3 mutations and PCD by six pathogenic genes (DNAAF3, DNAH5, CCDC103, CCDC114, CCDC40 and DNAH11) were the most frequent genetic disorders in the GLD group. For the GDL group, PID was the most common genetic disorder, in which the most enriched genes were BTK, CYBB, IL2RG and STAT3, followed by MM/S, where the most commonly mutated genes were CHD7, FBN1, COL2A1 and PTPN11. Additionally, a genotype–phenotype relationship and age distribution of genetic disorders are shown in detail in online supplemental figure S2.

Causative gene distribution among genetic lung disease and genetic disease with lung involvement. (A) Causative gene distribution of genetic lung disease; (B) causative gene distribution of genetic disease with lung involvement. ACDMPV, alveolar capillary dysplasia with misalignment of pulmonary veins; CCHS, congenital central hypoventilation syndrome.
GLD and GDL exhibit different clinical outcomes and medical management
Notably, 51.4% (72 of 140) of the patients had changes in clinical management after precise diagnosis in the following four areas5: (1) redirection of care (12.1%, 17 of 140), including adopting palliative care and termination of treatment; (2) initiation of new subspecialist care (13.6%, 19 of 140), including specialist care and other specific phenotypic monitoring; (3) changes in medication or diet (24.3%, 34 of 140); and/or (4) major procedures (7.9%, 11 of 140), including umbilical cord–blood stem cell transplantation and hematopoietic stem cell transplantation. The most common medical impact of molecular diagnosis was in improving ongoing management by changing treatment or diet (24.3%) and the initiation of new subspecialist care (13.6%) (figure 3). Following genetic diagnosis, significantly more patients with GLD had a change in medical management compared with those with GDL (19 vs 53, 82.6% vs 45.3%, p=0.001) (table 2).

{kind=link}
{kind=link}
{kind=link}
Impact of genetic diagnosis on medical management between patients with genetic lung disease and genetic disease with lung involvement.
Among patients with GLD, five of seven patients with SMD (P003, P36–38, P86) harbouring SFTPC mutations received long-term hydroxychloroquine treatment with informed consent for off-label drug use; three of them responded well at follow-up visits, showing catch-up of growth and withdrawal of oxygen supply; however, one patient died of disease exacerbation within 1 month after hydroxychloroquine treatment and the others did not respond to the treatment.6 In contrast, the other two patients eventually died of refractory respiratory distress syndrome (RDS) and critical illness without hydroxychloroquine treatment. Similarly, a pair of full-term twins (P001–002) died of progressive aggravation of RDS, which may have been due to homozygous ABCA3 mutations that were revealed following subsequent gene sequencing. Nine patients with PCD with mucociliary clearance disorders and three patients with CF with chloride channel dysfunction enabled new specialised nursing practices, including airway clearance through daily chest physiotherapy, coordinated inhaled hyperosmolar agents, and prompt and aggressive administration of inhaled tobramycin (300 mg nebulised two times per day); the latter was implemented for a 28-day period upon the first evidence of Pseudomonas aeruginosa growth, per a protocol that had been previously described.7 Furthermore, long-term oral administration of azithromycin was performed to protect patients (P084, P076) from respiratory infection and to ameliorate the decline in lung function.8 During follow-up visits for more than 6 months, there was no disease progression on imaging, and initial indications of improved lung function were revealed (data not shown).
As for the GDL in 53 patients, the primary impacts on medical management following molecular diagnosis included changes in medication and/or diet, especially for patients with PID. In contrast to patients with loss-of-function mutations in STAT1 and STAT3 sequences who received supportive therapy (eg, intravenous immunoglobulin) and antiviral/antimycobacterial prophylaxis, three patients harbouring STAT3 gain-of-function mutations with multisystem autoimmunity symptoms had interferon-gamma treatment and supportive therapy with long-term antifungal treatment.9 Two patients with activated phosphatidylinositol 3-kinase δ syndrome 1 (PIK3CD, P115–116) received rapamycin (Akt-mTOR signalling inhibitor) treatment, whereas two patients with STING-associated vasculopathy (TMEM173, P068 and P140) were subjected to ruxolitinib (JAK1/2 inhibitor) or tofacitinib (JAK1/3 inhibitor) treatment on an off-label basis.10 11 It is also crucial to monitor the emergence of severe phenotypes, such as prevention and monitoring of joint damage and ocular lesions of type-II collagenopathies (COL2A1, P030–31), multisystem comorbidity including kidney and eye lesions, and cancer risk of Rubinstein-Taybi syndrome (CREBBP, P072).12–14 Fortunately, 7 of the 11 patients (10 patients with PID and 1 patient with IMD) who had received transplantation achieved near-complete remission; four patients died of transplant-related complications, and additional two patients with SBDS and CYBB mutations were waiting for transplantation.
Moreover, for the additional 27.1% (38 of 140) patients, actionable medical measures could have been administered if an earlier molecular diagnosis or longer follow-up had been achieved (online supplemental table S2). We regretfully report that 33.6% (47 of 140) of patients died despite positive response to treatments, due to critical conditions/deterioration or the guardian choosing to give up treatment; 14 of them died before receiving molecular diagnoses that could have accounted for their deaths (table 2). It is also noteworthy that prenatal diagnoses of amniocentesis were ongoing in two families based on the probands’ genetic findings (P052 and P065).
Discussion
The present study represents the first comprehensive survey on the genetic spectrum of monogenic diseases with predominant respiratory involvement. Our data revealed differences in spectra of genetic disorders, clinical features and precise treatments/prognoses across age. Previous studies have provided detailed analyses on a variety of diseases, especially neurological phenotypes, such as intellectual disability, developmental delay and epilepsy.3 15–17 Additionally, some studies have investigated the overall genetic diagnostic rates in specialised wards or key laboratories.5 18–21 To the best of our knowledge, the median diagnostic yield of genome and exome sequencing of previous studies was approximately 33.2%, as determined by aggregate analyses, which highlights the clinical value of genetic sequencing as a diagnostic tool for paediatric patients.22 The overall diagnostic yield ranged from 8.4% to 68.3% in previous next-generation sequencing (NGS) research reports.23–27 In particular, a previous study showed a 13% diagnostic rate in the neonatal intensive care unit (NICU); the study also found that respiratory dysfunction was one of the phenotypes with the lowest molecular diagnostic rate in a comparative analysis of phenotypes in children.28 Our present study had similar results, with a 14.4% diagnostic rate.
Patients with MM/S accounted for 45.2% of patients in the neonatal group, significantly more compared with the infant and children groups (p<0.05). Unfortunately, 64.3% of patients with MM/S in the neonatal group ultimately died prior to the completion of the study. In a previous study that explored genetic causes of death in NICUs, MM/S was identified as the leading genetic cause of death.29 However, in our study, apart from MM/S, we found that neuromuscular disease and IMD also represented high death rate categories, of which the IMD was without obvious visual clues to genetic conditions. Early gene detection in these patients with non-specific clinical manifestations is crucial for establishing precise diagnoses and predicting prognoses.
Our data showed that four patients presenting refractory RDS as newborns, caused by ABCA3 and SFTPC mutations, had poor prognoses, and exogenous surfactants or systemic steroids were ineffective in treating them; these phenomena are consistent with those reported in the previous studies.30–32 Five of six infants/children with interstitial lung disease caused by SFTPC mutations received hydroxychloroquine treatments; three of them exhibited significant improvements in oxygen demand and chest CT.6 33 Since the prognosis of SMD varies greatly with phenotype and age as well as the effect of different gene mutations on protein function, it is important that SMD is precisely diagnosed as early as possible. It is especially important for neonates suffering from severe dyspnoea immediately after birth and in those with persistent or recurrent episodes in the absence of obvious systemic inflammatory condition. Notably, only three patients received the diagnosis of CF in our cohort, while it is by far the most common monogenic cause of respiratory disease in the white population. The low incidence of CF in Chinese children may explain our finding of very few patients with CF; of course, a possibility of missed detection due to lack of neonatal screening for CF also remains.
Certain entities of GDL, including PID, IMD and MM/S, which underlie typical respiratory phenotypes such as recurrent respiratory infections, have been highlighted in general paediatric practice.34 35 Furthermore, in the present study, 70 patients (63.9%) received the diagnosis of PID in the infant and children groups, which is much higher percentage compared with the results from previous studies.36–38 This discrepancy may be related to our role as a national tertiary referral chest centre.
Molecular diagnoses are of great importance for improving clinical management and family care strategies. However, they had a proportionally larger effect on GLD than on GDL (82.6% vs 45.3%, p=0.001). Moreover, 74% of patients with GLD obtained changes in medical care and medication; in contrast, only 24% of patients with GDL had changes in medical care and medication. On the one hand, in addition to specific treatment, pulmonologists have a greater focus on the respiratory management, which was most responsible for this difference; this includes regular airway clearance by combinations of physiotherapy and exercise, aggressive respiratory infection prevention, routine monitoring of pulmonary function, and periodical imaging, most of which have achieved substantial gains in disease stability and improvement in patients with certain monogenic disease.8 39 However, the focus from other clinicians on the airway management is far from sufficient compared with that on the origin of primary disease because the airway as the widest portal provides the most extensive exposure to the outside environment and contributes to exacerbation of the primary diseases to a certain extent. On the other hand, patient attitudes and compliance to respiratory management depend on their physicians. Thus, positive management of airway is of paramount importance until effective treatments for respiratory impairment emerge.
To the best of our knowledge, the present report represents the first large-scale NGS study to investigate the genetic spectrum and diagnostic yield, as well as the impact on clinical management of monogenic diseases in children with a predominant respiratory phenotype. Since the application of NGS is relatively underused in paediatric respirology, our present study further clarifies the importance of NGS in this field. Our findings demonstrated that more than 50% of NGS-diagnosed patients experience changes in their ongoing clinical management, and even more patients may benefit from such NGS diagnoses in terms of improving clinical management in the future.
Conclusions
Taken together, our findings demonstrated that the spectra of monogenic diseases in children with a predominant respiratory phenotype varied with age. Furthermore, half of the patients experienced a change in management following genetic diagnosis; there was a difference in change in clinical management and prognostic prediction between GLD and GDL. For such patients, precise diagnosis plays a crucial role in the formulation of treatment strategies and prognostic evaluations.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplemental information.
Ethics statements
Patient consent for publication
Ethics approval
This prospective, observational cohort study was approved by the Ethics Committee of the Children’s Hospital of Fudan University (approval number: 2014-121).
Acknowledgments
We thank all of the patients and their families for their participation in this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Research conception and design—LQ. Patient collection and follow-up—YC, XW and LW. Genetic testing, bioinformatics analysis and drafting the report—BW, HW, YL and XD. Genetic counselling—LY and BW. Data acquisition—DD, MM and LH. Data analysis and interpretation—all authors. Drafting of the manuscript—DD. Critical revision of the manuscript—LQ. Receiving grant—LQ. Approval of final manuscript—all authors.
Funding This work was supported by grants from the National Natural Science Foundation of China (No. 8167060762) and Shanghai Science and Technology Commission’s Scientific and Technological Innovation Action Plan (No. 16411950700).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.