Article Text

Abstract
Background Musculoskeletal complications of Down syndrome (DS) are common but infrequently reported. The combination of ligamentous laxity and low muscle tone contributes to increased risk of a number of musculoskeletal disorders and a delay in acquisition of motor milestones. The primary aim of this study was to describe musculoskeletal anomalies reported in a national cohort of children with DS.
Methods This was an observational study. Children with DS, aged 0–21 years, were invited to attend a musculoskeletal assessment clinic conducted by a paediatric physician. Relevant musculoskeletal history and clinical findings were documented.
Results Over an 18-month period, 503 children with DS were examined (56% male). The median age was 8.1 years (0.6–19.2). Pes planus was almost universal, occurring in 91% of the cohort. A range of other musculoskeletal anomalies were observed, with inflammatory arthritis (7%) and scoliosis (4.8%) occurring most frequently after pes planus. Delay in ambulation was common; the median age to walk was 28 months (12–84).
Conclusion Children with DS are at increased risk of a number of potentially debilitating musculoskeletal problems. These conditions can present in variable manners or be completely asymptomatic. Pes planus is common; therefore, early consideration of orthotics and lifelong appropriate supportive footwear should be considered. Delayed ambulation is frequently noted. A significant proportion of children with DS have arthritis; however, despite a high prevalence, it is often missed, leading to delayed diagnosis. An annual musculoskeletal assessment for all children with DS could potentially enable early detection of problems, allowing for timely multidisciplinary team intervention and better clinical outcomes.
- musculo-skeletal
- rheumatology
- syndrome
- orthopaedics
- comm child health
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
What is already known on this topic?
Joint laxity is almost universal in children with Down syndrome.
Hypotonia is a well-described feature of Down syndrome.
Children with Down syndrome are at increased risk of a number of musculoskeletal disorders.
What this study adds?
Foot, spinal and hip pathologies are likely underestimated.
Children with Down syndrome are at increased risk of inflammatory arthritis, which if undetected and untreated can lead to permanent joint damage and disability.
Musculoskeletal assessment should be part of the normal annual health surveillance programme for all children with Down syndrome.
Background
Down syndrome (DS) is a chromosomal disorder caused by full trisomy 21 (94%), mosaicism (2.4%) or translocations (3.3%). Ireland has an estimated incidence of 1 per 546 live births per year, the highest in Europe.1 The worldwide incidence is 1 per 1000–1100.2
Joint laxity, which may be associated with delayed ambulation, is thought to be almost universal in children with DS. This, combined with hypotonia, also a well-described feature of DS, has significant and widespread functional impact, and contributes to increased risk of a number of musculoskeletal disorders, a delay in acquisition of motor milestones and lower levels of physical activity in children with DS.3–6
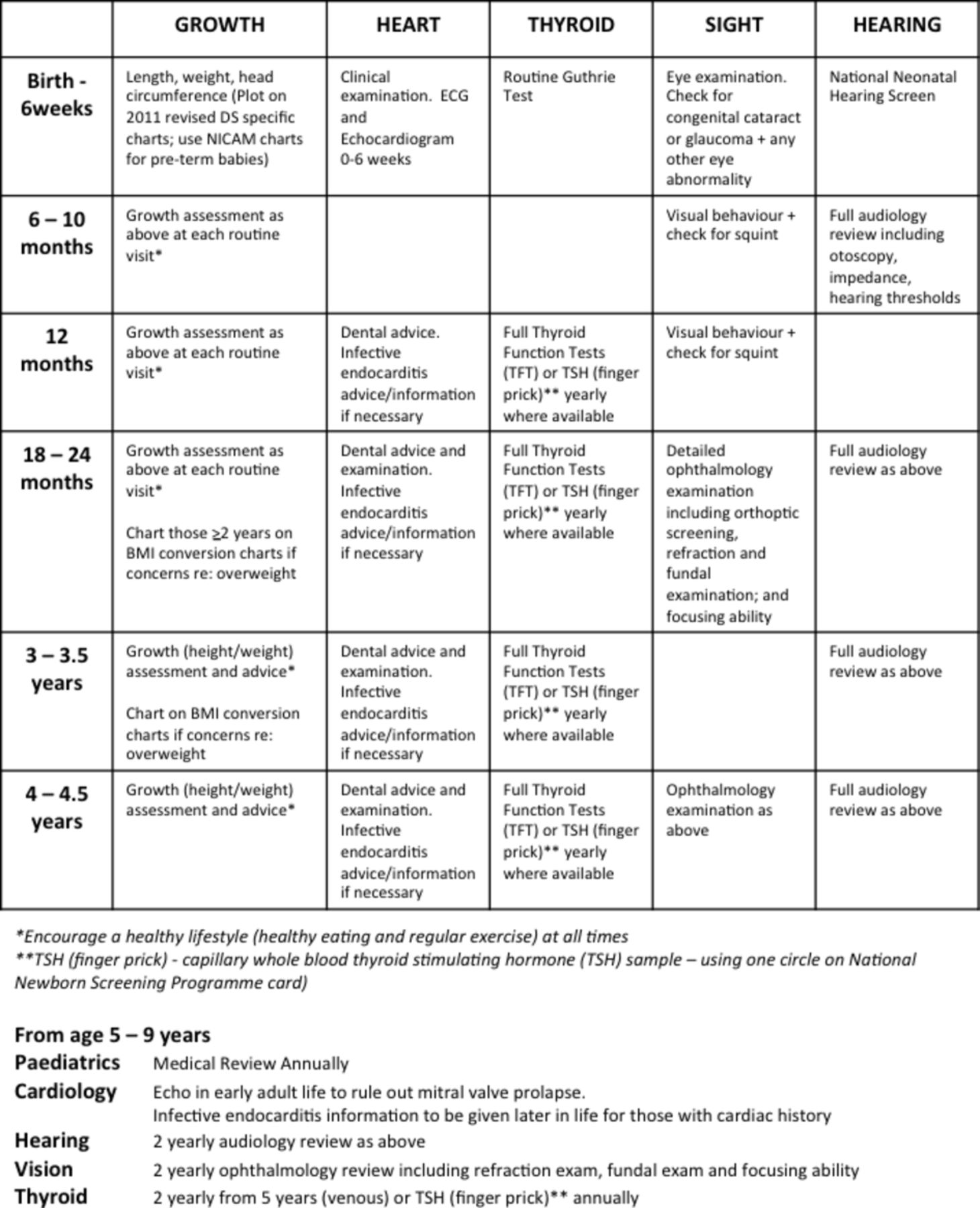
Medical management guidelines recommending scheduled health checks for children with DS include screening of growth, heart, thyroid, sight and hearing (figure 1).7 These guidelines have been reviewed and approved by national interest groups in Ireland and the UK (Down Syndrome Medical Interest Group (DSMIG)).

Down syndrome (DS) medical management guidelines. Suggested schedule of health checks taken from guidelines. BMI, body mass index; NICAM, Neonatal Infant Close Monitoring chart.
There remains a paucity of data with regard to the musculoskeletal features of DS, with inconsistent and variable recommendations for clinicians on appropriate screening and management of musculoskeletal anomalies. There appears to be a disproportionate focus on cervical spine (C-spine) instability, with little if any detail on other musculoskeletal conditions that occur more frequently in children with DS.
Objectives
To describe musculoskeletal anomalies reported in a national cohort of children with DS.
To document the presence or absence of hypermobility in DS.
Study design and methods
Type of study
This was an observational study carried out over an 18-month period.
Study population
The target population was children aged 0–21 years with DS living in Ireland. Using EUROCAT (European Surveillance of Congenital Anomalies) data,8 this was estimated to be 1652 children. The target population was sourced with the help of DS Ireland (1300 children registered with their service met the target population criteria). Other avenues used to source participants were general paediatric, specialist and developmental clinics attended by children with DS in the largest children’s hospital in Ireland, Our Lady’s Children’s Hospital, Crumlin; through liaison with the Chartered Physiotherapists in Intellectual Disability of Ireland; and by enrolment of the study in the Irish Paediatric Surveillance Unit. Children with DS were invited to attend local musculoskeletal screening clinics.
Sampling frame
Convenience sampling was used for the purpose of this study. Awareness and information about the study were made available to as many as possible of the target population through the avenues outlined above. Information for this report was gathered as a secondary outcome from a larger study where the primary objective was to identify inflammatory arthritis in children with DS.
Study assessments
Screening assessments took place nationally. At these clinics a comprehensive history was ascertained and a musculoskeletal examination performed by a paediatric doctor (PD). Hypermobile joints were assessed using the 9-point Beighton Hypermobility Score.9 There is no universal agreement on a paediatric threshold for joint hypermobility. For the purpose of this study, hypermobility was defined as a Beighton score ≥4.9 10 Concerns regarding joint inflammation and/or new musculoskeletal findings were further assessed at a second clinic. This was attended by the PD and a second examiner, a consultant paediatric rheumatologist (PR). Orthopaedic expertise was requested as required.
Inclusion criteria
All children with DS aged 0–21 years who responded to the invite to attend a local musculoskeletal screening clinic were assessed and included in the study. Information on musculoskeletal anomalies in children with DS was gathered from the reported medical history. Any child with a musculoskeletal anomaly detected through musculoskeletal examination at a screening clinic was further assessed by the supervising PR to confirm the initial findings. If uncertainty persisted, appropriate imaging was requested, and if required referrals to other specialties were made.
Exclusion criteria
Non-responders or those aged over 21 years at the time the condition occurred were excluded.
Results
Over an 18-month period, 503 children with DS were examined, 56% were male, with a median age of 8.1 years (0.6–19.2). Prenatal diagnosis of DS occurred in 10%; 15% were born preterm. The genotype of the cohort included 92% full trisomy 21, 5% mosaicism, 1% Robertsonian translocation, and 2% of parents were unsure, presumed full trisomy 21.
Musculoskeletal anomalies and DS
The almost universal finding in our cohort was pes planus (91%) (figure 2A). This was recorded in children who demonstrated loss of the medial longitudinal arch of their foot. Twenty-four per cent of these children did not avail of orthoses. Figure 2B reflects the wide range of other musculoskeletal anomalies identified.
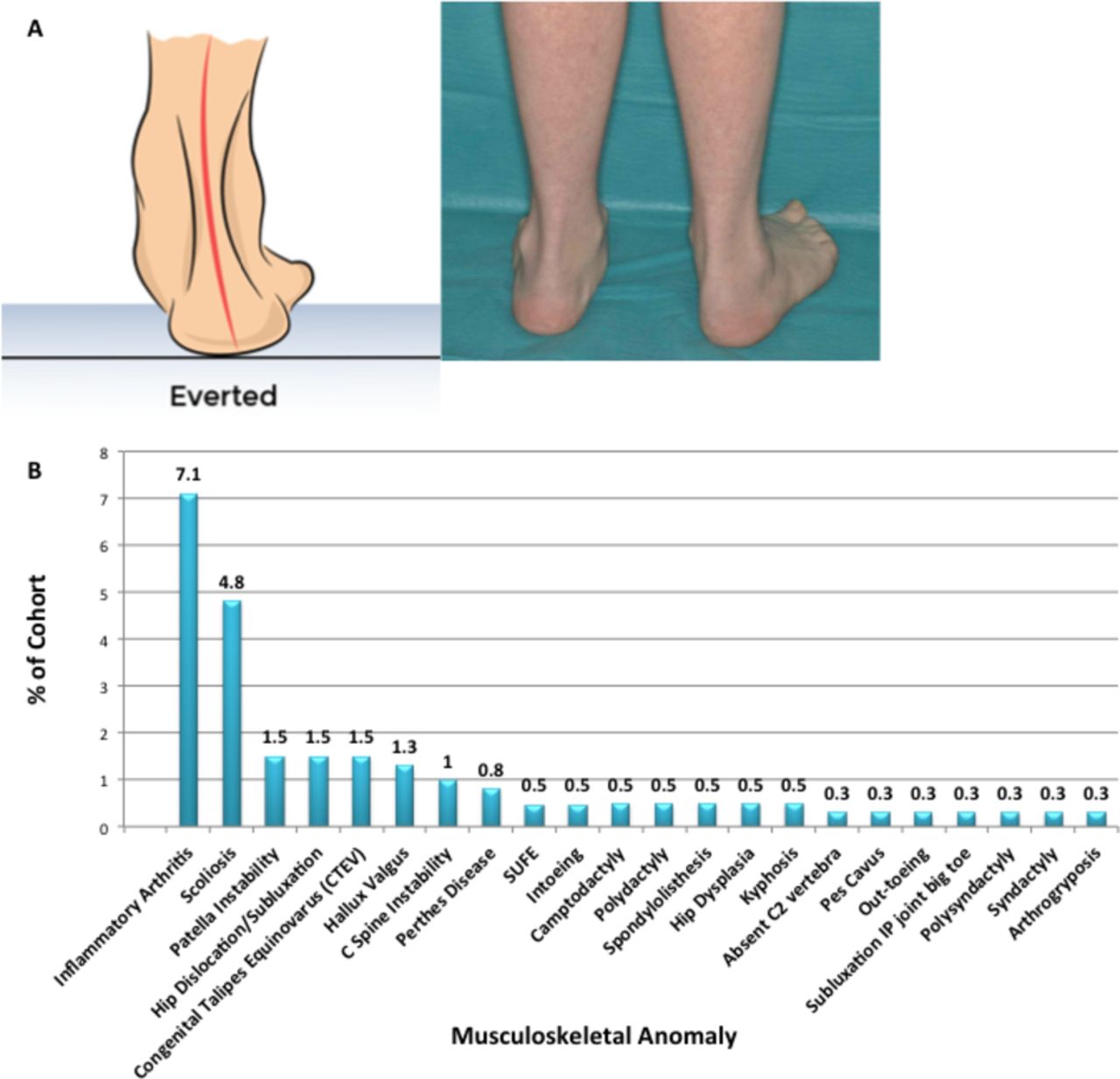
Musculoskeletal anomalies detected in a cohort of children with Down syndrome. (A) Pes planus, highlighting the inability of the calcaneus to come out of eversion in a child with Down syndrome. (B) Range of other musculoskeletal anomalies reported in our cohort. SUFE, slipped upper femoral epiphysis. IP, interphalangeal.
Inflammatory arthritis was diagnosed in 7% of children assessed, confirmed following examination by both the PD and PR. Scoliosis, confirmed clinically or radiologically, occurred in 4.8%. Other spinal abnormalities identified included C-spine instability (1%), spondylolisthesis (0.5%) and absent C2 vertebra (0.3%). Hip pathologies reported included dislocation and subluxation (1.5%), Perthes disease (0.8%), slipped upper femoral epiphysis (0.5%) and dysplasia (0.5%).
Ambulation and DS
The median age the cohort walked was 28 months (12–84 months).
Hypermobility, Beighton score and DS
The average Beighton score for the cohort was 4; the average score of adolescents (defined as children >12 years; 27% of cohort) was 2 (0–6), significantly lower than that recorded for children, 4 (0–9) (p<0.001). Just over half (59%) of those examined scored ≥4, with 2% scoring the maximum of 9 points (figure 3A). Using the Beighton scoring system, the criteria that scored most often (figure 3B) were hyperextensibility of the little finger metacarpophalangeal joint (70% scored 1 or 2 for this criterion) and the thumb to forearm manoeuvre (53% scored 1 or 2 for this criterion). Notably, a high proportion of children had hypermobility of the hips and ankles (defined as greater than expected range of movement for age 11), joints not accounted for using the Beighton criteria.

Application of Beighton scoring system to a cohort of children with Down syndrome. (A) Percentage of cohort per Beighton score. (B) Percentage of cohort per Beighton score component.
Discussion
Musculoskeletal complications of DS
Hypotonia is well described in children with DS and is usually related to the skeletal muscles. Type VI collagen is crucial for skeletal muscles. Collagen α1 (VI) and α2 (VI) chains are encoded by genes located on chromosome 21 and are expected to have higher dosage in individuals with DS.12 The combination of ligamentous laxity and low muscle tone contributes to an increased risk of a number of musculoskeletal disorders, such as C-spine instability, hip instability, scoliosis and foot problems.3 4
A delay in acquisition of motor milestones and lower levels of physical activity in children with DS have been observed.5 Reduced physical activity contributes to the development of lower bone mass, obesity, and a failure to develop or maintain maximum possible muscle strength.13 14 Inappropriately low expectations of physical activity and motor function from family, healthcare workers and self feed into this cycle. Conversely, however, overattributing motor difficulties to low tone and hypermobility may lead to missed pathology and misdiagnoses.15 16
The foot and DS
Pes planus (figure 2A) is common and was almost universal in our cohort (91%). Young people with pes planus and incorrect footwear are at risk of callus formation over pressure points, repetitive ligamentous injury and development of bone spurs.6 17–21 Inability of the calcaneus to come out of eversion (figure 3A) leads to hindfoot valgus, resulting in multiple postural changes. This almost certainly contributes to inability of many children with DS to sustain good strength when they stand or build good core musculature. Digital deformities also occur, such as hallux valgus.
Expertise of a paediatric podiatrist should be acquired routinely to allow for timely detection and management of foot deformities. Podiatric interventions and good footwear will help to encourage a more ‘normal’ foot posture, in turn improving posture and quality of life, which are underestimated and neglected.22 23 In severe cases, orthopaedic review and surgical intervention may be required.
The spine and DS
C-spine instability is a well-described orthopaedic condition associated with DS. It involves either the occiput-C1 level (atlanto-occipital instability) or the C1–C2 level (atlantoaxial instability).
C-spine instability on plain film was present in 1% of our cohort. This is significantly lower than previous studies that estimate 10%–27% of individuals with DS may have radiological findings of instability. However, most are asymptomatic, with only 1%–2% developing symptomatic instability.24–28
Low correlation between radiological findings and symptoms emphasises the need for a high index of suspicion if symptoms develop. Commonly, the signs and symptoms of symptomatic C-spine instability progress over time and include neck pain, abnormal head posture, torticollis, reduced neck movements, deterioration of gait and/or frequent falls, increasing fatigability on walking or deterioration of manipulative skills. In the presence of any of the above, appropriate history, examination and investigation should be undertaken. Complications from spinal cord compression can worsen suddenly. While serious complications are rare, death can occur.24
A review of the recommendations for C-spine screening reminds us there have been years of controversy and discussion. Currently there is no screening procedure that can predict those at risk. In particular C-spine X-rays in children have no predictive validity for subsequent acute dislocation/subluxation at the atlantoaxial joint.25–29 In an attempt to reduce morbidity and mortality from C-spine instability, the DSMIG developed a basic surveillance essential for people with DS. They suggest that routine radiological screening for asymptomatic people with DS is not recommended. Asymptomatic individuals with DS should not be barred from normal sporting activities because there is no evidence that participation in sports increases the risk of C-spine injury any more than for the general population.30 31 For specialised sport, such as gymnastics, children with DS should not be automatically excluded, but the requirements of national governing bodies which include a clinical screening protocol should be observed.32
Assessment for scoliosis should be routine and ongoing in all children with DS. Our study identified that just under 5% had either clinical or radiological evidence of scoliosis. Vigilance is required in particular for those that may be at risk of thoracic curves secondary to early thoracotomies from congenital heart surgery. Spondylolisthesis may also occur and present with the classic symptoms of low back pain and radiculopathy.
The hip and DS
Hip instability in DS is common, with incidence reported to be between 1% and 7%. The natural history is often progressive, with the typical onset of hypermobility of the hip evolving to habitual dislocation, persistent subluxation and fixed dislocation. Without appropriate intervention this can eventually lead to loss of independent mobility.33 Our study identified that 1.5% had hip dislocation or subluxation.
Radiographic studies demonstrate that, in comparison with a normal acetabulum, the acetabulum of an individual with DS is deep, more horizontally placed and has increased anteversion, while the proximal femur has a normal neck-shaft angle and a moderate increase in anteversion.34 This in association with generalised hypotonia likely increases the risk of hip abnormalities in DS.
The most common presenting sign for hip pathology is a limp, which may often be painless. Hip X-rays should therefore always be performed in a child with DS presenting with a limp.
The knee and DS
Patella instability has been reported to occur in DS to varying degrees (1.5% in our cohort), from mild subluxation to complete dislocation. Altered gait may be a sign of patella instability and a decreased range of motion of the knee may be noted. Orthoses or even corrective surgery may be required.
Inflammatory arthritis and DS
Children with DS are at increased risk of developing inflammatory arthritis. In 1984, Yancey et al35 were the first to describe seven children with DS having a juvenile idiopathic arthritis (JIA)-like arthropathy, classified as having ‘Arthropathy of DS’ (A-DS). There are no published population surveys establishing the prevalence and incidence rates of A-DS, but crude estimates suggest the incidence is threefold greater than JIA. Prevalence has been estimated to be 8.7 per 1000.16 36 In our cohort, the prevalence (20 per 1000) is significantly greater than previously reported. Despite higher incidence and prevalence rates, A-DS is often under-reported and/or misdiagnosed. A polyarticular pattern of disease, with predominance in the wrists and small joints of the hands, is most commonly seen (figure 4).

Arthropathy of Down syndrome (A–DS). Child with A-DS demonstrating the ‘prayer sign’. Highlights restriction in wrist extension secondary to inflammatory arthritis. Also note the swelling of the metacarpophalangeal and proximal interphalangeal joints bilaterally.
The average time to diagnosis of A-DS in our cohort was 1.7 years (0.2–4.9), significantly longer than reports for JIA.37 As a result of delayed diagnosis and therefore delayed instigation of appropriate treatment, children with A-DS are presenting with significant joint damage and disability at diagnosis. Bone erosions were evident on X-ray in 42% of our cohort at diagnosis, significantly higher than observed in a JIA comparison group (erosions 14%, p<0.0001).
Reasons for delayed diagnosis are multifactorial, including poor verbal skills and altered pain expression. It has been reported that children with DS express pain more slowly and less precisely than a child without DS.38 Children with A-DS often adapt to pain with reported observations such as slowing mobility, reluctance to hold a parental hand or behavioural change. When assessing a child with DS, careful consideration of possible interpretations of the history and a thorough clinical examination will aid correct and timely diagnosis of A-DS. Signs and symptoms can be subtle, so a high index of suspicion should be employed. MRI with gadolinium contrast may be required if there is clinical uncertainty.
Ambulation and DS
Significantly delayed ambulation is noted in children with DS. Our results with regard to acquisition of walking unaided are comparable with the literature that reports children with DS walk at 23 months (13–48), compared with 13 months (9–17) for the general paediatric population.39 Early multidisciplinary team intervention is important to ensure these children obtain their full potential with regard to acquisition of walking unaided.
Hypermobility, Beighton score and DS
In our cohort of children with DS, Beighton scores appear to underestimate the level of hypermobility observed during clinical examination. In fact, in our cohort, adolescents (ie, age >12 years) were not hypermobile (average Beighton score of 2), and children only barely in the hypermobile range, using the Beighton scoring system. We observed a high proportion of children had other joints that extended beyond the normal range of motion, in particular hips, ankles and small joints. There is a mismatch of the scored joints using the Beighton system and those most likely to be hypermobile in DS (figure 5). The Beighton score also fails to identify the severity of hypermobility characteristic in these children. A condition-specific hypermobility scoring system would address these issues.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Joints included in the Beighton Hypermobility Score. (B) Most frequently hypermobile joints within the study cohort of children with Down syndrome.
Conclusion
Musculoskeletal anomalies in DS are common and underestimated. In particular, there is a significant risk of inflammatory arthritis. We estimate that the prevalence of A-DS is 20 per 1000, with a phenotype much more erosive and aggressive than previously described. This observational study highlights the importance of regular musculoskeletal screening to allow timely detection and management of these treatable conditions. We advocate that children with DS have an annual musculoskeletal assessment as part of their health surveillance programme.
The limitation of this study design is that the musculoskeletal evaluation of children was primarily to assess whether there was evidence of inflammatory arthritis. Information on a significant number of the musculoskeletal anomalies was gathered from the medical history only; therefore, occurrence of some conditions might in fact be underestimated. Due to the sampling frame employed, bias may be introduced into the results as parents with musculoskeletal concerns may have been more inclined to accept the invite to attend for assessment.
Acknowledgments
Special thanks to Down Syndrome Ireland and the children and families that participated in the study.
References
Footnotes
Contributors CF and OGK: substantial contributions to the conception or design of the work; acquisition, analysis or interpretation of data for the work; drafting the work or revising it critically for important intellectual content; final approval of the version to be published; agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding This work was supported by the National Children’s Research Centre, Crumlin, Dublin (grant number D/13/4 5).
Competing interests None declared.
Patient consent Not required.
Ethics approval Ethical approval for this study was granted by the Ethics (Medical Research) Committee Office, Our Lady’s Children’s Hospital, Crumlin, Dublin, Ireland.
Provenance and peer review Not commissioned; externally peer reviewed.