Article Text

Abstract
Referral for an assessment of tall stature is much less common than for short stature. Although the commonest cause is an underlying familial tendency to tallness, there are important disorders that should be considered at the initial assessment. Distinguishing these conditions from normal variations of growth is the key objective when managing the child and family. In some children, further targeted investigations will be needed and in rare instances intervention to limit final height may be appropriate. This article discusses a structured approach to the assessment and management of a child with tall stature.
- Endocrinology
- General Paediatrics
Statistics from Altmetric.com
Introduction
At the population level, similar numbers of children have a height greater than 2 SD above the mean as those who have a height 2 SD below the mean. Despite this, a referral for the assessment of tall stature is much less common than a referral for short stature, highlighting societal perceptions of what is and what is not acceptable. Most tall children are healthy and simply have a familial tendency to tallness but there are a number of important pathological causes that need to be considered. Children who are tall in relation to their parents, that is, taller than expected given parents’ heights but with a height SDS<2 may also require investigation. Hence parental size, as well as population standards, needs to be taken into account. Occasionally, intervention may be necessary to limit height, and in some children identifying the underlying diagnosis will lead to the introduction of a tumour surveillance schedule.
Normal prenatal and postnatal linear growth
Fetal growth is mediated by insulin, insulin-like growth factor (IGF)-1, IGF-II and maternal nutrition, and is independent of endogenous growth hormone (GH) secretion. Recent evidence suggests that firstborns are taller than later-born children, with an incremental height reduction from first to third birth order.1 Increasing paternal age at childbirth has also been associated with taller stature and a slimmer phenotype in girls.2 It is possible that birth order influences imprinted growth genes such as IGF-II and H19, and in some animal studies firstborns have increased GH and IGF-1 receptor responsiveness.3 ,4
Postnatal linear growth can be divided into three phases: infancy, childhood and puberty. In each phase, the predominant influence on linear growth is different. During infancy, nutritional status is the main influence whereas the GH–IGF-I axis is the major regulator of linear growth during childhood. Sex steroids have a major impact on the growth pattern during puberty.
In the first 2 years of life, a period of ‘catch-up’ or ‘catch-down’ growth commonly takes place as individuals establish a growth trajectory that they then follow until puberty. Abnormal growth can be explained on the basis of alterations in factors impinging on the growth plate or intrinsic changes in the growth plate itself.
Constitutional advancement of growth (CAG) describes a normal growth pattern that is the mirror image of constitutional delay of growth and puberty (CDGP). Children with CAG have accelerated growth after birth reaching their peak centile by 2–4 years of age, and then the child grows along this centile till puberty, which is often early.5 This is in contrast to the child with CDGP who grows slowly and progresses into puberty late (figure 1). The CAG growth pattern is distinct from familial tall stature where children are born with an above average birth length. Growth is normally rapid during early childhood, but slows by around 4–5 years of age, after which the growth trajectory of the healthy tall child runs parallel to the relevant height centile until puberty. The healthy 4–5-year-old child will not cross height centiles at this stage of childhood.
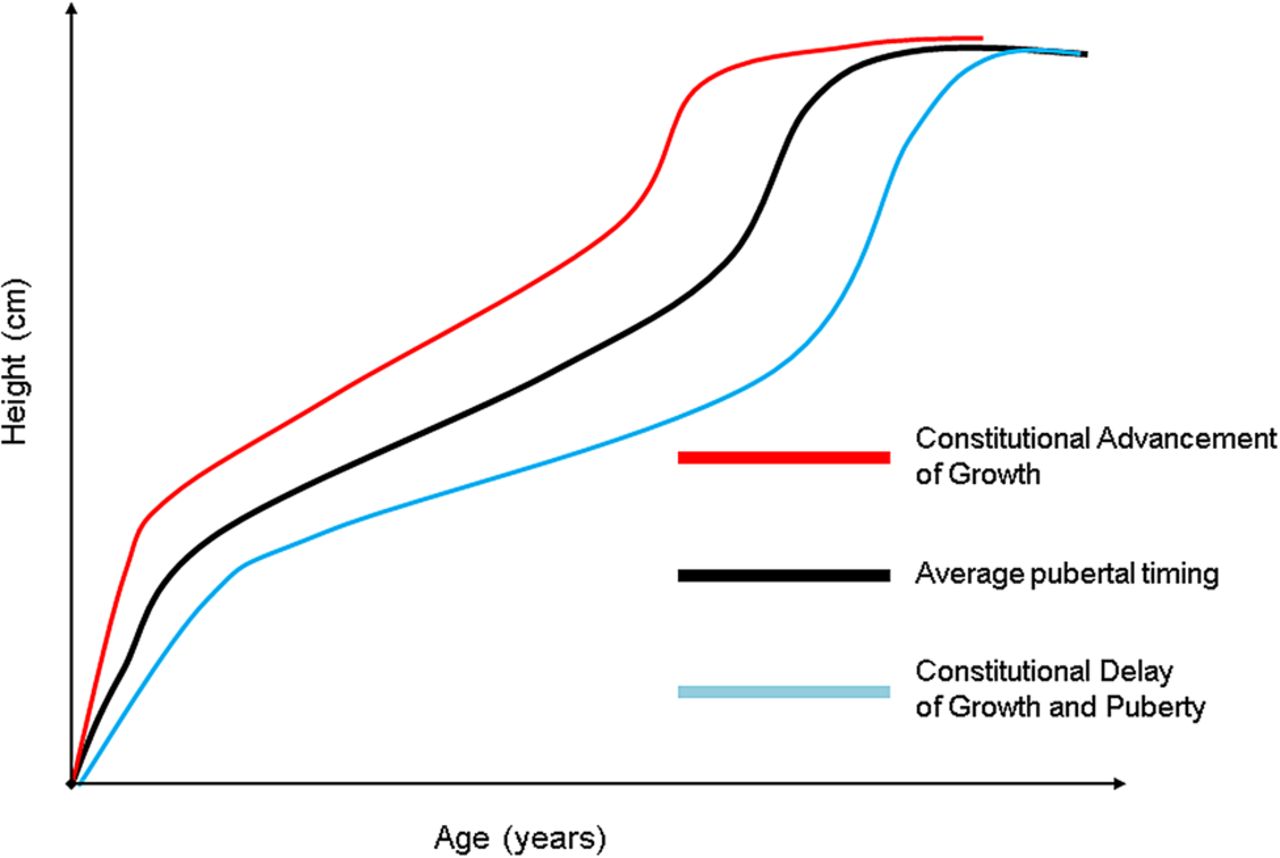
Schematic representation of the growth patterns of constitutional advancement of growth and constitutional delay of growth and puberty.
There may be a minor acceleration in linear growth during mid-childhood associated with the onset of adrenarche. The pubertal growth spurt reflects increasing endogenous sex steroid production and associated increased GH secretion. In girls, the growth spurt starts approximately 2 years earlier than in boys, and the onset coincides with the start of breast development. In girls, peak height velocity occurs at an average of 12 years of age and is followed by the onset of menarche after which height velocity decelerates with approximately 2 years growth (around 5 cm) left till final height. In boys, the onset of the pubertal growth spurt occurs when testicular volumes are 6–10 mL and peak height velocity occurs at a testicular volume around 12 mL at an average of around 14 years of age. Most children are not ‘average’ and will progress through puberty earlier or later than this general description.
General considerations when assessing the child with tall stature
A detailed history and physical examination will exclude the need for investigation in most cases. It is important to exclude obesity and a Body Mass Index (BMI) should be calculated from the height and weight then plotted on a BMI chart. It is essential to assess the pubertal status to exclude precocious puberty and evaluate thyroid status to exclude hyperthyroidism. Visual fields and fundoscopy should be assessed together with head circumference.
A sitting height measurement is useful to assess disproportion and when considering the differential diagnosis of tall stature (figure 2). A subischial leg length is calculated by subtracting the sitting height from the standing height. The sitting height and subischial leg length measurements should be plotted on a sitting height/subischial leg length chart. These measurements can be converted to an SD score to calculate the SD subischial leg length less SD sitting height value, which will give an indication of the degree of disproportion. More specific measurements may be indicated such as arm span if Marfan's syndrome (MFS) is suspected.
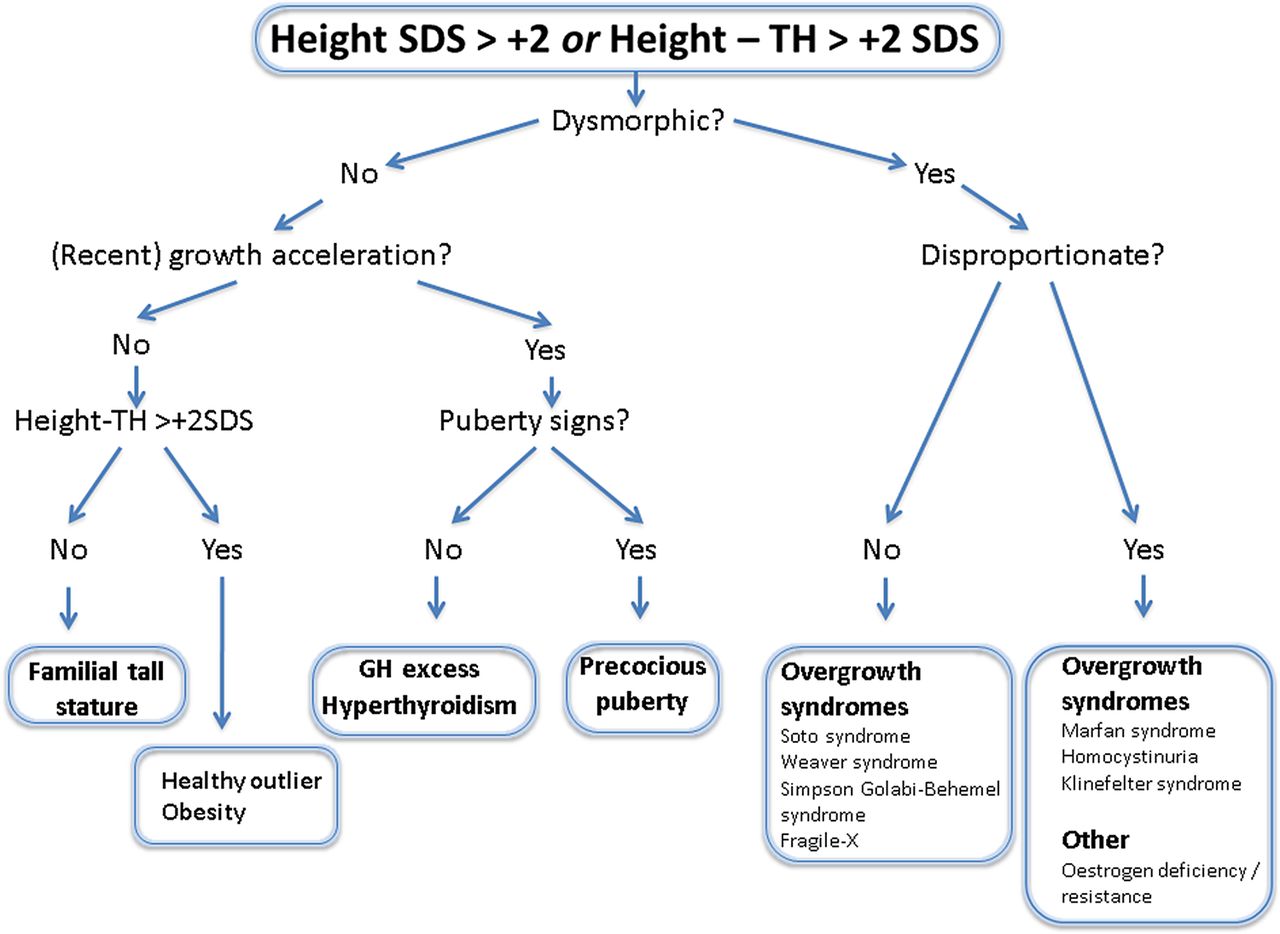
Diagnostic approach to tall stature.8 Target height (TH). TH is defined as: Boy's=(father's height+mother's height)/2+7 and girl's=(father's height+mother's height)/2–7.
Measurement of parental heights is needed to calculate the target centile range and estimate height prediction. Measured rather than reported parental heights are preferable as adults may overestimate their height. A key consideration is the fact that around 10% of healthy children will still fall outside the target centile range. Furthermore, mid-parental height performs less well as predictor of adult height when parental heights are at the extremities of the height range.6 Serial height measurements are needed to evaluate height velocity.
Specific considerations when assessing the tall child
When considering the differential diagnosis of tall stature, it is helpful to consider the diverse mechanisms leading to excessive growth (boxes 1 and 2).7 The causes will reflect perturbations in the normal growth process as outlined above: an abnormal growth plate, excess nutrition in early life, alterations in the GH–IGF-I axis and changes in sex steroid action.
Classification of tall stature6
-
Normal variant
-
Familial tall stature
-
Constitutional advancement of growth
-
-
Excess or modulation of growth factors
-
IGF-1, eg, obesity
-
Insulin, eg, obesity
-
IGF-II, eg, Beckwith–Wiedemann syndrome
-
-
Excess sex steroid production
-
Gonadotrophin-dependent precocious puberty
-
Gonadotrophin-independent precocious puberty
-
-
Deficiency of factors needed to arrest linear growth
-
Hypogonadotrophic hypogonadism
-
Aromatase deficiency
-
Oestrogen receptor deficiency
-
-
Deficiency of factors needed to prevent bone elongation
-
Marfan's syndrome
-
Homocystinuria
-
-
Excess of growth gene
-
SHOX (Klinefelter syndrome)
-
Y-specific control gene
-
-
Excess growth hormone secretion
-
Pituitary tumour – somatotropinoma
-
McCune–Albright Syndrome
-
Multiple endocrine neoplasia type 1
-
Carney complex
-
-
Excess or mutation of growth factor receptors
-
Trisomy IGF-1R
-
Inactivating mutation of FGFR3
-
Overgrowth syndromes associated with tall stature
-
Marfan's syndrome
-
Beckwith–Wiedemann syndrome
-
Sotos syndrome
-
Weaver's syndrome
-
Marshall–Smith syndrome
-
Homocystinuria
-
Simpson–Golabi–Behmel syndrome
A well, tall child with a normal examination and tall parents is likely to have familial tall stature but the apparently well prepubertal child may still have an underlying chromosomal abnormality. Obesity from overnutrition may cause tall stature and calculating a BMI and then plotting on a BMI chart to calculate the SD score will be useful in subsequent discussions with the family. Sex steroid excess from gonadotrophin-dependent and gonadotrophin-independent precocious puberty will result in accelerated linear growth and tall stature during childhood.
While a GH-secreting tumour can result in tall stature in childhood, disorders that impair sex steroid production or metabolism will usually result in tall stature in adolescence and early adulthood—a time when sex steroids are normally important variables in the growth process. These rare disorders include aromatase deficiency, oestrogen resistance and androgen insensitivity.
The clinical evaluation will determine the subsequent investigations (box 3 and figure 2)8 and an assessment by a geneticist may be merited in some cases. As trisomy X (XXX female) and an XYY male may have tall stature as the only clinical feature, a karyotype is indicated to detect these conditions. Serum homocysteine levels should be measured in children with mental retardation or marfanoid features. As fragile X is a frequent cause of learning difficulties, DNA testing for fragile X should be considered. If MFS is suspected, then a cardiac assessment that includes echocardiography with assessment of aortic root dimensions should be obtained.
Investigations for tall stature
-
Karyotype
-
T4 and TSH
-
IGF-1
-
Bone age
-
If concerns about puberty consider:
-
LH, FSH, oestradiol or testosterone
-
Consider AFP, HCG in precocious puberty
-
-
If marfanoid features consider:
-
Plasma homocysteine
-
DNA for FBN1 mutation
-
-
Store DNA if tall stature syndrome suspected
Absent or delayed puberty causes tall stature in adolescence and beyond because of delayed epiphyseal closure and prolonged prepubertal growth. Basal gonadotrophins and sex hormone levels should be measured in those with an abnormality of pubertal development. An individual with a female phenotype can occasionally have a 46XY karyotype with the associated tall size that an XY karyotype imparts.
Selected causes of tall stature
Familial (constitutional) tall stature
This is the commonest reason for referral for tall stature and is usually evident by 4 years of age. Girls are more frequently referred but overall the numbers presenting are falling which probably reflects greater social acceptance of tall stature. Parental anxiety is common, especially in girls and if the mother was similarly affected.
There is relatively high GH secretion and IGF-1 levels compared with average or short children.9 The physical examination is normal and the presenting height is usually within the target centile range calculated from the parent's heights. An increase in height velocity may be observed in early childhood until about 4 years of age and is normal thereafter. A bone age may be advanced and may be used for adult height prediction. The parents and child should be reassured that the growth pattern is normal although a period of growth monitoring may help to reassure those involved prior to discharge.
Obesity
Obese children are taller than expected during childhood but final height is not usually increased. There is an increased predisposition to adrenarche, a relatively early puberty, hyperinsulinism and an advanced bone age, all of which contribute to increased height velocity and taller stature during childhood. Those with early puberty have reduced subsequent linear growth compared with normal weight controls resulting in normal adult height.10
Klinefelter syndrome
Klinefelter syndrome (XXY) is the commonest sex chromosome aneuploidy causing tall stature. The prevalence is approximately 1 in 500 to 1 in 1000. The phenotype is variable but the clinical features frequently include tall stature (due to increased leg length), variable learning difficulties and behavioural problems. Individuals usually progress through puberty spontaneously but have relatively small testes (less than 8 mL in volume) with biochemical evidence of gonadal failure by late adolescence and subsequent infertility. Exogenous testosterone supplementation is usually appropriate. In some cases surgery for gynaecomastia is indicated.
Marfan's syndrome
The main features of this autosomal dominant disorder are tall stature, ectopia lentis and aortic root dilatation and a fibrillin-1 (FBN1) gene mutation is found in the majority of cases. Recently, the Ghent criteria for diagnosis have been modified to avoid overdiagnosis of MFS and the recognition of two overlapping conditions, the MASS (mitral valve prolapse (MVP), aortic enlargement, skin and skeletal findings) phenotype and MVP syndrome (box 4 and table 1), which do not carry the same risk factors for aortic dissection and aneurysm associated with MFS.11 The new scoring system emphasises two important features of MFS, aortic root aneurysm/dissection and ectopia lentis, as well as molecular genetic testing of the FBN1 gene. If there are insufficient systemic features (<7), a borderline aortic root dilatation (<3 SDS) without FBN1 mutation, the term ‘non-specific connective tissue disorder’ can be used initially. If an FBN1 mutation is identified in a sporadic or familial case but aortic root dilatation is <3 SDS, the term ‘potential MFS’ has been proposed.11
Revised Ghent criteria for diagnosis of MFS11
In the absence of family history:
-
Ao≥2 SDS and EL=MFS*
-
Ao≥2 SDS and FBN1=MFS
-
Ao≥2 SDS and systemic score≥7=MFS*
-
EL and FBN1 with known Ao=MFS
In the presence of a family history of MFS:
-
EL AND FH of MFS (as defined above)=MFS
-
Systemic score≥7 AND FH of MFS (as defined above)=MFS
-
Ao≥2 SDS above 20 years of age, Ao≥3 SDS below 20 years of age +FH of MFS (as defined above)=MFS
*Exclude Shprintzen–Goldberg syndrome, Loeys–Dietz syndrome and Ehlers–Danlos syndrome.
Ao, aortic diameter at the sinuses of Valsalva or aortic root dissection; EL, ectopia lentis; FBN1, fibrillin-1 mutation; MFS, Marfan's syndrome; SDS, SD score.
Scoring of systemic features11
Homocystinuria
Patients with homocystinuria have similar clinical features to MFS but with associated learning difficulties, an increased predisposition to psychiatric disorders and a thromboembolic tendency. It is an autosomal recessive disorder due to mutation in the cystathionine β-synthase gene and the diagnosis is indicated if plasma homocysteine levels are elevated.
Overgrowth syndromes
Sotos syndrome
Individuals with Sotos syndrome have distinctive facies (prominent forehead, hypertelorism, downslanting palpebral fissures, pointed chin), large dolicephalic head, excessive growth and learning difficulties. There is prenatal and postnatal overgrowth and disproportionately long limbs. The height velocity is increased during the first 4 years of life and is normal or height-appropriate thereafter. Adults are tall with some reaching excessive heights, but most do not require intervention to limit final height.12 There may be delayed motor development, delayed speech and learning difficulties, although up to 10% have no reduction in IQ. Deletions in the NSD1 gene account for the majority of cases and there may be autosomal dominant transmission.
Weaver syndrome
This syndrome is also characterised by prenatal and postnatal overgrowth, typical facies (hypertelorism, large ears, depressed nasal bridge, downslanting palpebral fissures, broad face, dimpled chin, prominent wide philtrum, micrognathia), hypo or hypertonia, advanced skeletal maturation and camptodactyly. Many have developmental delay and learning difficulties. A mutation in the EZH2 gene is causative and occasionally there is autosomal dominant transmission.
Endocrine causes of tall stature
Precocious puberty
Sex hormone excess, from either gonadotrophin-dependent or gonadotrophin-independent causes, may lead to accelerated linear growth and tall stature. Sex hormones act on the growth plates to cause maturation of chondrocytes and longitudinal bone growth. Although children may be tall or relatively tall for family size, there is advanced skeletal maturation with premature fusion of the epiphyses and final height will usually be compromised in the absence of intervention.
Sex hormone deficiency or resistance
Sex hormone deficiency or resistance leads to delayed fusion of the epiphyses with prolonged prepubertal linear growth and tall stature in adolescence and beyond. There will be long legs and low upper segment ratio (eunuchoid proportions). Notable causes of sex hormone deficiency include hypogonadotrophic hypogonadism. Sex hormone resistance from aromatase deficiency or mutations of the oestrogen receptor α gene is extremely rare.
GH excess
Excessive GH synthesis is very rare in children and is usually caused by a GH-secreting pituitary adenoma (somatotropinoma) or hyperplasia of the somatotrophs. Associations include MEN-1 syndrome, Carney complex and McCune Albright syndrome. Tall stature and/or accelerated linear growth are observed in children prior to closure of the epiphyseal growth plates and acromegalic features (such as enlarged jaw, hands and feet) may be observed in adolescence. Puberty may be delayed due to the pressure effects on the pituitary gonadotropes. There may be false positive results in normal tall children investigated with a GH suppression test (carbohydrate ingestion will usually suppress GH secretion) and IGF-1 levels must be interpreted with an age-specific reference range and may be elevated during puberty and in taller children.13 About 50% of somatotropinomas cosecrete prolactin. An MRI with pituitary-specific views should be performed in suspected cases. Treatment strategies include GH receptor antagonists, somatostatin analogues, dopamine agonists, trans-sphenoidal surgery or radiotherapy.
Other endocrine causes
Hyperthyroidism will cause accelerated linear growth and advanced skeletal maturation and tall stature. Familial glucocorticoid deficiency is a rare autosomal recessive disorder characterised by glucocorticoid deficiency, elevated adrenocorticopic hormone and normal mineralocorticoid production and tall stature.
Interventions aimed at limiting final height prognosis
The majority of children will require no intervention to limit height and an explanation and reassurance will suffice. In certain cases, however, the option of intervening should be discussed as children may become so tall that it impacts on their quality of life. In this scenario, the expectations of the family should be explored and the potential complications from intervening should be discussed in full. Height prediction algorithms have been devised to predict final height in the otherwise healthy child and may be a useful component of the discussion.14 ,15 Unfortunately, the SD associated with a given prediction is usually considerable and this tends to hamper their value.
There are two main management strategies: high dose sex steroid administration and epiphysiodesis.
High dose sex steroid administration
Administration of high dose oestradiol or testosterone has been used to limit final height prognosis by promoting epiphyseal closure. However, their use has declined over the last 20 years because of side-effects during treatment.15 High dose oestradiol may cause weight gain, nausea, benign breast disease, excessive vaginal discharge, pigmented areolae ovarian cysts and thrombosis, whereas high dose testosterone may cause acne and gynaecomastia. Of concern is the fact that girls treated with high dose oestrogen appear to have an increased incidence of compromised fertility in adulthood with a depleted follicular pool and the development of primary ovarian insufficiency.16 This effect appears to be dose-dependent with higher doses more likely to cause problems.17 To date, high dose testosterone treatment has not been shown to affect male fertility in adulthood18 although long-term follow-up studies have not been undertaken to evaluate whether there are other potential complications. Serum homocysteine levels should usually be measured before oestrogen treatment is administered to limit final height.
Some have advocated inducing puberty by the use of more physiological doses of sex steroid before the tall child progresses into puberty as a means of reducing final adult height.19 The rationale for this approach is that the return from the pubertal phase of growth is limited and the sooner sex steroid is introduced, the more likely it is to impact on final adult height. This strategy can be compared with the impact of early or precocious puberty on adult final height. Unfortunately, there will be a phase when treatment will make the already tall child even taller and discordant from their peers in pubertal status as well as height.
There remains a significant amount of uncertainty regarding the efficacy of exogenous sex steroids in limiting final height whether used as part of a high or lower dose strategy.
Epiphysiodesis
Epiphysiodesis is a surgical intervention undertaken under general anaesthetic by a paediatric orthopaedic surgeon that destroys the growth plates around the knee (distal femur, proximal tibia and fibula) using a drill and curettage, thus preventing further longitudinal bone growth. In very tall boys, the use of bilateral percutaneous epiphysiodesis around the knee has been reported to reduce final adult height by 5 cm when undertaken at a mean bone age of 13.9 years20 and there was normalisation of body proportions. Others have recommended that the intervention should be performed at a bone age not exceeding 12.5 years in girls and 14 years in boys, and that a treatment decision should be taken before girls have reached a height of 170 cm and boys 185 cm. A potential side-effect is the development of leg length discrepancy although in a recent series this was not observed.21 This intervention remains controversial and further data on its safety profile are required.
Strategies to slow growth in pituitary gigantism
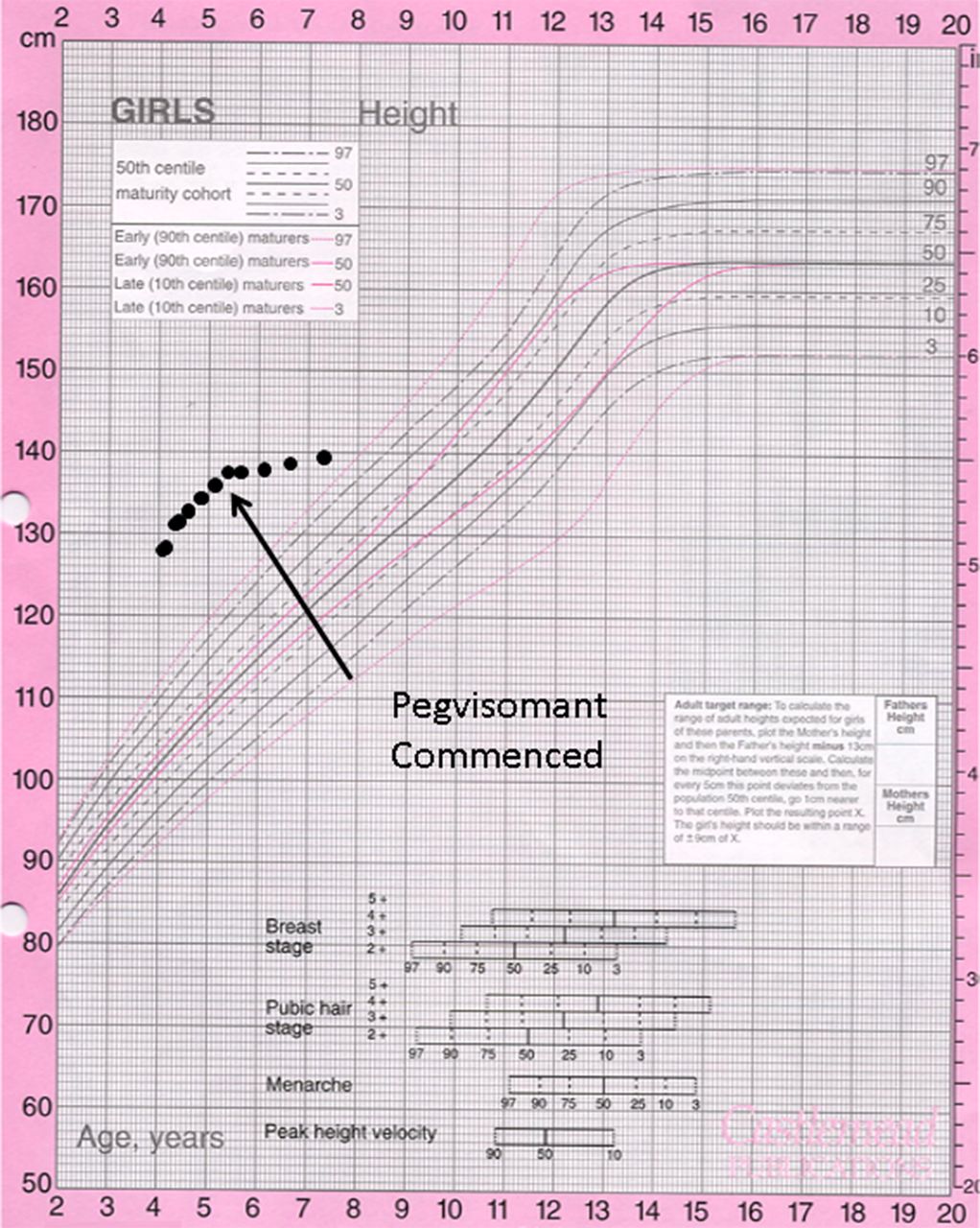
Medications that have been used to reduce the excessive growth in pituitary gigantism include octreotide and pegvisomant. Both are administered by subcutaneous injection. Octreotide, a somatostatin analogue, suppresses GH production but has significant gastrointestinal side-effects and can cause glucose intolerance. Pegvisomant is a selective GH receptor antagonist that binds to the GH receptor but prevents signal transduction. It is a highly effective way of reducing growth velocity in the patient with pituitary gigantism (figure 3) but cannot be recommended in other settings.

{kind=link}
{kind=link}
{kind=link}
Growth chart of a child with pituitary gigantism before and after treatment with pegvisomant. Note the excessive tall stature at presentation.
Summary
Many children referred because of concerns regarding tall stature have familial tall stature and require no intervention, but the family and child may still require support and reassurance. There are a number of diagnoses that should be considered at the initial assessment, as additional investigations and surveillance may be necessary. In rare instances, intervention to limit height may be merited following a full discussion of the potential risks and benefits.
References
Footnotes
-
Contributors The first draft was prepared by JHD with subsequent input/revisions from both authors.
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.