Article Text

Abstract
Paediatricians need to develop a strategy for assessing and managing the short child because it is a common reason for referral to paediatric services. Understanding what is normal is a key prerequisite to the appropriate assessment of the short child. Most pathological causes of short stature will be associated with clues in the history or on examination. Factors that should trigger a more detailed assessment of the short child include malaise, dysmorphic features, slow growth and small size with a normal weight centile. Establishing that the healthy short child is growing appropriately for their family size can be reassuring for the family and clinician and will facilitate discharge.
- Growth
- General Paediatrics
Statistics from Altmetric.com
Introduction
Only a minority of short children, which can be defined as a height less than 2 standard deviation score (SDS) below the mean (less than the 2nd centile), will be found to have an underlying pathology. A diagnosis can frequently be reached without the need for further investigation, and this article will focus on some of the key features that indicate that a child is well and can be discharged. It will also focus on some of the pointers towards underlying pathology and introduce a framework that can be used to ensure that the assessment and plan of investigation are comprehensive. The same framework can also be used to explain the rationale for the assessment and diagnosis to the family. A clinical case will illustrate how the described framework can be used.
Initial assessment
The further away from the population mean the child's height lies, the more likely it is that they will have an underlying pathology. It has been estimated that around 1 in 5 children with a height less than 2 SD below the mean (2nd centile) and around half the children with a height less than 3 SD below the mean will have a pathological reason for their small size.1 The exact figure will depend upon the nature of the population studied and the reference standards used. Similarly, the slower a child is growing, the more likely it is that they will have an underlying pathology. The child's current height (length in the case of the infant) together with earlier measurements should be plotted on appropriate reference charts and be further placed into context by comparing this with the parental target. The likelihood of pathology in the short child of shorter parents will be different to a child of the same size who has taller parents. Many contemporary charts have details of how to do this but, essentially, a point that is half-way between the mother's and father's heights on the centile charts (ideally measured rather than reported) is used to generate a range between which most of the couples’ children can be expected to lie. The phenomenon of ‘regression to the mean’ whereby short parents tend to have children who are not as short as they are should also be borne in mind.2 Some standards include a correction that takes this into consideration when calculating the parental target. It is helpful to show the growth chart to the child and parents and to explain how the child's height compares with the population and to that expected given parental size.
There are a number of fundamental questions to ask as part of the initial assessment in addition to establishing who it is that is concerned in the first place. Establishing the child's birth weight will help to indicate whether short stature could be linked to poor growth in utero (was the child small for gestational age at birth?). By the age of 4 years, around 10% of children born with a low birth weight (<−2 SDS or the 2nd centile) will stay short with no substantial ‘catch-up’ growth. Babies with extremely low birth weight are particularly at risk of this growth pattern. Finding out whether the child is well or not is clearly important although there may be discrepancy between what the child thinks and what parent(s) think.
Linear growth and stature in early life
Infants can cross height (or more precisely length prior to 2 years of age) centiles for physiological reasons in the first 2–3 years of life, and this, coupled with the difficulties associated with obtaining accurate measurements in the very young child, can make interpreting growth data challenging at this time. Nutrition is one of the key determinants of growth during infancy, and nutritional status will be a central component of any growth assessment. Growth hormone (GH) becomes a major determinant of height velocity as the child matures. Children with isolated GH deficiency have a weight and length that is similar to the normal population at birth,3 but by the time the child is 2 years old, they will be short.
Assessment beyond infancy
Beyond infancy, and before puberty, the healthy child will grow parallel to the reference centiles. Obese children are typically taller than expected based upon mid-parental height, and it follows that the obese child will therefore have become tall at a relatively early age. The obese child who is below predicted height may have a growth disorder. Chronic illness can sometimes impact more on weight than height, but endocrine disease, such as isolated GH deficiency, will primarily affect height centile rather than weight, and so, comparing the two (height centile and weight centile) is helpful. A key component of the assessment will be a history and examination that targets features of chronic illness and dysmorphic syndromes. A particularly difficult component of the assessment is to gauge whether flaws in the nurturing process could be affecting growth and nutrition. An abusive environment (physical, emotional or neglect) is a well-recognised cause of poor growth and short stature although there may be little in the way of clues in the outpatient department. Extreme behaviour with features, such as soiling and food hoarding have been described,4 but the more subtle end of the spectrum is challenging to diagnose.
Puberty
Puberty and the associated increase in sex steroid production can make life more difficult for the paediatrician because it introduces another variable that requires consideration and assessment. Relatively early or late puberty may be associated with centile crossing on height charts, and so height velocity cannot be used as an index of health or disease in the same way as it can be in the prepubertal child. The growth spurt is an early feature of puberty in girls but is a relatively late feature in boys where there is progressive gonadotrophin production and associated testicular enlargement for some time without an increase in growth rate. The rise in height velocity at puberty is linked to rising oestrogen levels in both sexes and reflects an increase in pulsatile GH release together with a direct effect of sex steroid on the epiphysis. Delayed progress into puberty will be associated with delayed growth acceleration, and for a time the individual progressing into puberty late may therefore be considerably shorter than his or her peers. Assessing the short child requires an understanding of the Tanner pubertal staging system, and it is difficult to reach a diagnosis in the short teenager without knowing an individual's pubertal status. The combination of relatively late progress into puberty and short parents can result in a healthy child appearing very short for a while when compared with peers or when plotted on charts. The timing of puberty has a major inherited, genetic component, and establishing when the parents progressed into puberty (when was your menarche? were you a lot shorter than your peers for a time?) is important. Boys will typically grow around 20–30 cm following the onset of puberty, while girls will grow between 15 and 25 cm.
Short stature—particular considerations
Dysmorphic syndromes
Most paediatricians become skilled at identifying dysmorphic features, but it is worth becoming well frequented with the features of the commoner conditions, such as Turner syndrome, Noonan's syndrome and Silver–Russell syndrome. Prader–Willi syndrome is associated with hypotonia and poor feeding in infancy in stark contrast to the hyperphagia in later childhood. Disorders associated with short stature and for which there may be clinical clues include abnormalities of the gene SHOX (stature homeobox gene on the X chromosome) which result in a condition called dyschondrosteosis. This condition is of interest because it is inherited in an autosomal-dominant fashion and can easily be confused with familial short stature.5 SHOX is situated in the pseudoautosomal region of the sex chromosomes and it is haploinsufficiency of this gene that also results in the short stature of Turner syndrome. In Turner syndrome SHOX haploinsufficiency will occur in association with loss of part or all of an X chromosome. The key clinical features reflect the fact that SHOX is important in normal skeletal development, and include Madelung's deformity of the forearm (also seen in Turner syndrome) which may become more noticeable as the individual matures. Many skeletal disorders are inherited in an autosomal-dominant manner, and measuring the skeletal proportions of the child and potentially the parents as well is important. Charts that document sitting height and sub-ischial leg length are a useful resource in any general paediatric and growth clinic. Sitting height is measured with a specific stadiometer and then subtracted from height to generate sub-ischial leg length. A pronounced discrepancy between the two SD scores raises the possibility of an underlying skeletal dysplasia. Pseudohypoparathyroidism (PHP) is associated with small stature in adolescence and in adult life, but the clinical phenotype in early childhood may provide little in the way of obvious clues, because these children may be relatively tall at this stage of life, and because the phenotype is extremely varied.6 Children with PHP are more likely to be overweight, and measuring calcium and PTH concentrations may be important if a pathological cause of short stature is suspected.
Endocrine causes of short stature
Autoimmune thyroid disease that destroys the thyroid gland (atrophic thyroid disease) will result in a characteristic phenotype, slow growth and short stature (figure 1). GH deficiency is rare, and in classical form has a characteristic phenotype with mid-facial crowding, and a significant amount of overlying subcutaneous fat. The child with a well-defined abdominal musculature is unlikely to have GH deficiency. Cushing's syndrome will occasionally be due to an adrenocorticotrophic hormone-producing adenoma (Cushing's disease), but signs of glucocorticoid toxicity in childhood and adolescence, including slow growth and short stature, are more likely to arise in association with exogenous steroid (oral, inhaled, topical).

Twins, one of whom has atrophic thyroid disease (autoimmune thyroid gland destruction) which has resulted in slow growth and short stature. Reproduced from Minerva, BMJ 1998;317:1668.
Recent developments—abnormalities of the GH-IGF-1 axis
A more detailed understanding of the various components of the GH–insulin-like growth factor 1 (GH-IGF-I) axis has resulted in an appreciation that defects of the axis between hypothalamus and growth plate can result in short stature of varying severity. A normal birth weight is typical in disorders seen in the upper part of the GH-IGF-1 axis (eg, mutations in GH-1 gene), whereas, a history of low birth weight suggests a problem in the lower half of the axis (eg, mutations in IGF-1, IGF-1R genes). Isolated GH deficiency that is due to defects in the growth hormone releasing hormone (GHRH) receptor or GH1 gene are extreme examples of what is a continuum of underlying genetic and associated biochemical and phenotypic defects. The GH-IGF-I axis is detailed in figure 2 and haploinsufficiency of GH receptor (Laron syndrome), IGF-binding proteins, including the acid-labile subunit (ALS) and of the IGF-I receptor with associated short stature are described.7 The small size may be relatively subtle in the case of heterozygous mutations of growth hormone receptor (GHR), ALS and the IGF-1R with a stature that is not necessarily out-with the population norm. IGF-I gene deletions are extremely rare and typically result in a more profound phenotype with a height that is several SDs below the mean when both genes are abnormal.8
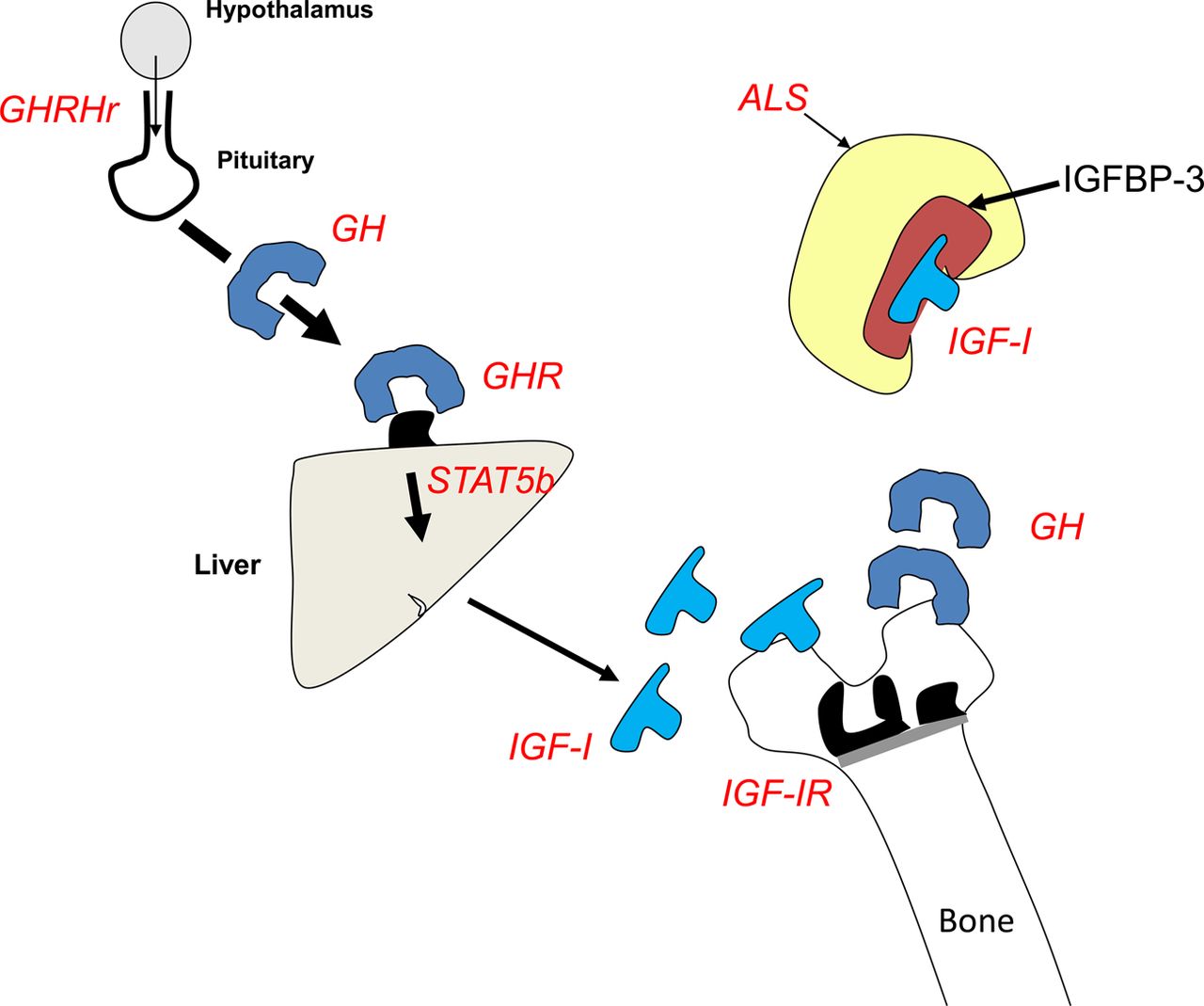
Some of the key components of the growth hormone (GH)/insulin-like growth factor 1 (IGF-1) axis highlighting a number of sites where gene defects (compound heterozygous/homozygous and in some instances heterozygous) can result in compromised growth and small stature. The text in red highlights the gene which, when mutated, can result in small stature. GHRHr refers to the GH-releasing hormone receptor (mutations of which can result in GH deficiency). Mutations of the GH gene itself can also result in GH deficiency. GHR refers to the GH receptor, ALS refers to the acid-labile subunit (part of the IGF-1 binding complex in serum) and STAT-5 refers to children with abnormalities of signal transducer and activator of transcription type 5b (involved in signalling ‘beyond’ the GHR). Children with IGF-1 (insulin-like growth factor 1) gene defects and IGF-1R (IGF-1 receptor) gene defects will also grow abnormally and be short as a result.
The consultation process
The ‘growth circle’ (figure 3) is a framework that can be used to facilitate the assessment of the short child. There is evidence that pictorial representations of information can lead to a greater understanding of the subject and better retention of information for future recall. We move clockwise from physiological to pathological causes of small size with the additional component represented by the uncommon endocrine causes of small size. It can act as an aide-memoir for the clinician and helps to educate families by categorising the reasons why children may be short in a systematic but pictorial manner. Working around the categories within the circle provides a forum for discussion including societal norms and the role of puberty before moving into the more pathological causes. Families involved in these discussions can follow the diagnostic process and see the rationale behind planned investigations. Recognition of the need for a karyotype to be checked, a chronic disease screen, pituitary function tests or referral to, as an example, the genetics team, usually follows naturally from these discussions. Our experience is that families find this kind of involvement in the diagnostic process useful, and our impression based upon follow-up consultations are that the retention of information is good.
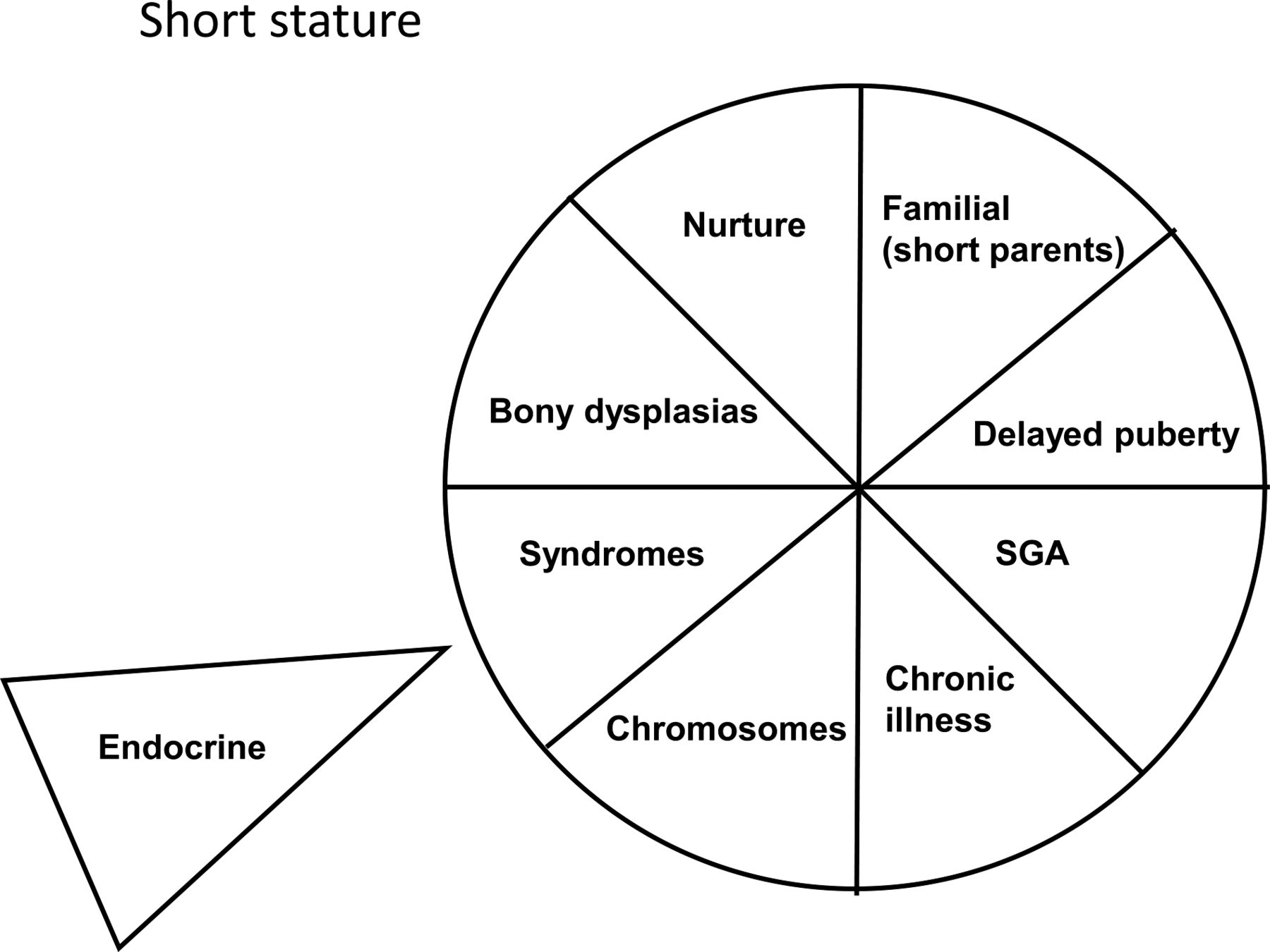
A summary of the various causes of small stature in childhood and adolescence in pictorial format.
Investigations
Blood
If there are questions about possible underlying chronic illness (eg, inflammatory or malabsorbtive conditions), an endocrine disorder, or indeed if there is simply unexplained small size, then the investigations in box 1 should be considered. If GH deficiency is suspected, then there are helpful guidelines that can be followed. GH deficiency results in a particular phenotype and growth pattern with low GH and GH-dependent factors on further assessment. Dynamic testing of the GH-IGF-I axis is only one component of the assessment of the child with suspected GHD,9 and should not be used in isolation as a means of establishing who should or should not receive GH therapy and especially if there are nutritional concerns (box 2).
Potential investigations in the child with unexplained short stature
-
Full blood count
-
Inflammatory markers (eryrthrocyte sedimentation rate, C-reactive protein)
-
Renal function (creatinine)
-
Bicarbonate (abnormal in, for example, renal tubular acidosis)
-
Hepatic function (can be abnormal in, for example, coeliac disease).
-
Coeliac screen
-
Thyroid function
-
Bone profile
-
PTH (may be abnormal in, for example, pseudohypoparathyroidism)
-
Insulin-like growth factor 1 (IGF-I) (a marker of growth hormone production which requires an age-related reference range).
-
Karyotype
Key features of growth hormone (GH) deficiency
-
History—low glucose in early life
-
Slow growth
-
Small stature
-
Dysmorphism with mid-facial crowding and central adiposity
-
Low GH dependent factors (insulin-like growth factor 1 (IGF-I), IGFBP-3)
-
Low GH on provocation testing
-
Abnormal imaging—for example, ectopic posterior pituitary bright spot
-
Rapid growth on GH replacement
Radiology
Bone age
Although not a diagnostic investigation, a bone age assessment is useful because it can indicate whether a child is likely to be taller with respect to their peers at final height (in which case it will tend to be delayed) or shorter with respect to their peers (in which case it will be advanced). Most children will, therefore, have either an advanced or delayed bone age. The ‘faster’ growers who progress into puberty early, and individuals who are tall will tend be in the former category, and the slow growers who progress through puberty late, or individuals who are below average height, will tend to be over-represented in the latter group. Bone age can be used to predict final adult height (with varying degrees of accuracy) but repeated, annual assessments are usually unnecessary unless it is going to provide additional diagnostic information or influence management. An advanced bone age in the short child should make one consider an underlying skeletal dysplasia.
Intracranial imaging
MRI of the hypothalamo-pituitary axis is important in all children with GH deficiency to exclude pathologies such as craniopharyngioma and germinoma.
Children with idiopathic GH deficiency frequently have an abnormal hypothalamo-pituitary axis on MRI with relatively subtle changes that include an ectopic posterior pituitary bright spot (figure 4). This is a very specific finding which is therefore not found in healthy individuals.
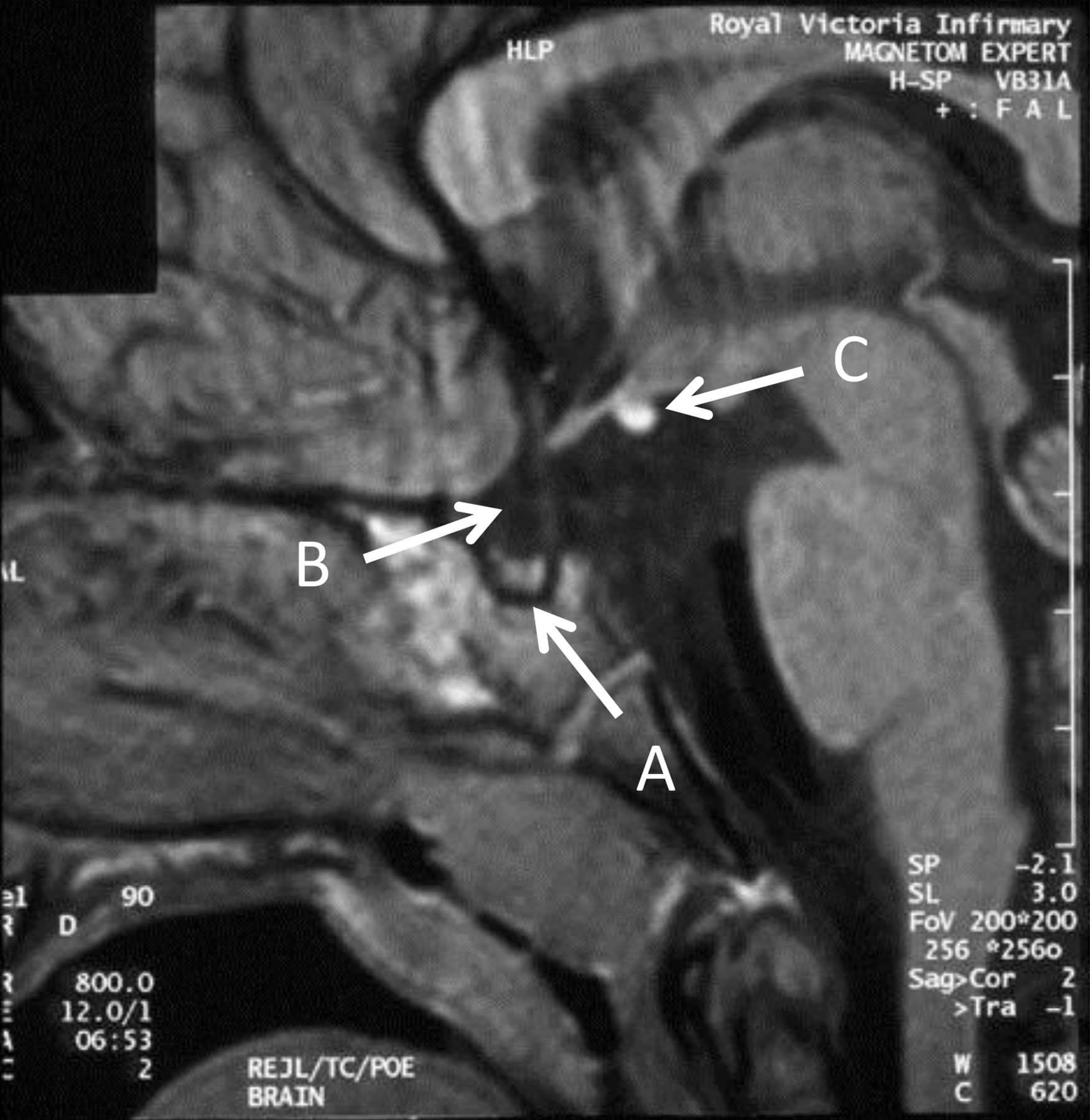
Cranial MR scan of patient with combined pituitary hormone deficiency. The key abnormalities to highlight are the small pituitary (A), poorly developed stalk (B) and ectopic posterior pituitary bright spot (C).
Management
Genetic short stature and individuals who are short because of late progress into puberty make up the vast majority of short stature referrals to paediatricians. It may be feasible to discharge at an early stage or following review in 6–12 months’ time, when an accurate measurement of height velocity can be used to confirm that all is well. When the cause of short stature is pathological rather than physiological then treatment will depend on the underlying cause. GH has been recommended as a growth-promoting therapy by National Institute of Health and Care Excellence (NICE) in a range of different disorders including children who are born SGA and who stay small at 4 years of age, and children with SHOX haplo-insufficiency.10 The impact of this treatment in the setting of short stature due to GH deficiency tends to be more profound than in the GH-sufficient causes. There should be a low threshold for involving the clinical genetics team where the child is dysmorphic or has prenatal and postnatal small size (box 3).
Notable causes of prenatal and postnatal small size
-
Congenital infection
-
Fetal alcohol syndrome.
-
Distal abnormalities of the growth hormone (GH)/insulin-like growth factor 1 (IGF-1) axis
-
Chromosomal copy number variant
-
Imprinting defect, for example, Silver Russell Syndrome
-
Confined placental mosaicism
-
Skeletal dysplasias—for example, 3M syndrome
-
Subtle syndromic diagnoses, for example, Floating Harbor
Case study
One of the twins in the picture (figure 5) has an underlying pathology. By the time children are around 2 years of age they find a centile and adhere to it, and so either twin 1 must be growing abnormally quickly or twin 2 must be growing abnormally slowly. Causes of rapid growth include the onset of puberty, thyrotoxicosis and GH excess. Twin 1 had none of these conditions, and so it must be twin 2 growing slowly. His slow growth cannot be physiological at this age (both boys are prepubertal) and it cannot reflect the fact that he was born small for gestational age—in which case he would always have been smaller. There is no suggestion of a syndromic diagnosis (he is not dysmorphic), or a bone dysplasia, and his disposition argues against either psychosocial growth failure or overt chronic illness. He does not look hypothyroid, and if he had idiopathic GH deficiency he would be short from early life and not taller than his brother. On the basis of this clinical assessment it can be argued that the key investigations include the exclusion of chronic illness (by assessment of his tissue transglutaminase (TTG) antibody status and creatinine—all of which were normal). He must, therefore, have an evolving cranial lesion that is affecting endocrine function and more specifically GH release. Imaging revealed that the child had a craniopharyngioma.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Twins, one of whom is growing slowly (twin 2). See text for further details.
Conclusions
Many paediatricians will be referred short children, and a diagnosis can frequently be reached without the need for any investigations. The child who feels well, looks well and who has a height that lies within the parental target can frequently be discharged. There will usually be clinical clues in the child with a pathological cause of small size such as malaise, dysmorphism or a height that is at odds with parental size. A simple ‘aide memoir’, such as that described above, can be used to explain the rationale for an assessment and management strategy to the family.
Key learning points
-
The further away from the population mean the child's height is then the more likely it is that they will have an underlying pathology.
-
The further away from the parental target height the child is, the more likely it is that they will have an underlying pathology.
-
Children with GH deficiency are not slim and do not have a well-defined musculature.
-
The short, heavy child is much more likely to have an underlying pathology than the relatively tall heavy child.
References
Footnotes
-
Contributors The first draft was prepared by TC with subsequent input/revisions from both authors.
-
Competing interests None.
-
Patient consent Obtained.
-
Provenance and peer review Commissioned; externally peer reviewed.