Article Text

Abstract
Background Noonan syndrome (NS), a relatively common autosomal dominant disorder with an incidence of 1 in 1000 to 2500 live births, is the most common syndromic cause of congenital heart disease after Trisomy 21.
Objective To comprehensively define the spectrum of cardiac morphology and specific clinical course of a large cohort of NS patients.
Design Retrospective, descriptive case series study.
Patients An international Harvard-based NS registry was combined with clinical data from NS patients followed at Boston Children’s Hospital, Massachusetts, USA.
Results We identified 293 patients with NS. Cardiovascular disease was seen in 81% (n=237) including pulmonary stenosis in 57%, secundum atrial septal defects in 32% and hypertrophic cardiomyopathy in 16%. A genetic mutation of the RAS-MAPK signalling pathway was identified in 62% (n=136). Genotype-phenotype associations were noted between PTPN11 mutations and atrial septal defects (p=0.001), and pulmonary stenosis (p<0.001). RAF1 mutations were associated with hypertrophic cardiomyopathy (p<0.001). Cardiovascular outcomes that differed specifically in a NS cohort included high re-intervention rates (65%) after percutaneous balloon pulmonary valvuloplasty for valvar pulmonary stenosis. Additionally, in NS patients with hypertrophic cardiomyopathy, a clinically significant regression of hypertrophy (17%) was observed as was a markedly higher incidence of concomitant congenital heart defects (70%).
Conclusions Patients with NS have a distinct spectrum of cardiac phenotypes that may have a natural history and response to therapy atypical to that normally seen in non-syndromic heart disease. A diagnosis of NS in a patient with pulmonary stenosis or infant-onset hypertrophic cardiomyopathy would facilitate condition-specific counselling on outcome and prognosis.
- Cardiology
- Genetics
- Syndrome
Statistics from Altmetric.com
What is already known
-
Noonan syndrome, an autosomal dominant disorder with variable expression, is the most common syndromic cause of congenital heart disease after Trisomy 21.
-
Valvar pulmonary stenosis, often with dysplastic valve leaflets, is the most common congenital heart malformation reported with an incidence of 50–60%.
-
Hypertrophic cardiomyopathy, found in 20% of patients with Noonan syndrome, has a worse risk profile at presentation compared to idiopathic or familial hypertrophic cardiomyopathy.
What this study adds
-
Noonan syndrome patients undergoing percutaneous balloon pulmonary valvuloplasty have a high re-intervention rate (65%) for residual valvar pulmonary stenosis.
-
Hypertrophic cardiomyopathy in Noonan syndrome is associated with a markedly higher incidence of concomitant congenital heart disease in 70%.
-
In the infant-onset cohort, spontaneous regression of hypertrophic cardiomyopathy was observed in 17% of affected Noonan syndrome patients.
Introduction
Noonan syndrome (NS) (OMIM 163950), first described in 1963, is a relatively common autosomal dominant disorder characterised by distinctive facial features including hypertelorism, ptosis and low-set ears; short stature; developmental delay of variable degree; lymphatic vessel dysplasia and cardiac defects (see figure 1).1–3 It is a genetically heterogeneous condition with an estimated incidence of 1 in 1000 to 1 in 2500 live births and is the most common syndromic cause of congenital heart disease (CHD) after Trisomy 21.4 ,5 Gain-of-function mutations in the RAS-MAPK signalling pathway are found in up to 60% of patients with NS with resultant perturbations in developmental processes including morphological determination, organogenesis and growth.6 ,7

Phenotypic features of Noonan syndrome (NS). (A) 14-month-old girl with NS characterised by tall forehead, hypertelorism, down slanting palpebral fissures, flat nasal bridge, and low set ears; (B) 7-year-old girl with NS characterised by curly hair, broad forehead, widely spaced eyes, and low set ears; (C) 5-year-old boy with NS characterised by widely spaced and down slanting eyes, flat nasal bridge, low set ears, peaked vermillion boarder of upper lip and pointed chin.
The incidence of heart disease in NS patients is reported to be as high as 82–90%.5 ,8–12 Patients with NS have been noted to have a suboptimal outcome to percutaneous balloon pulmonary valvuloplasty (PBPV) for valvar pulmonary stenosis (PS).13 Additionally, NS patients with a diagnosis of hypertrophic cardiomyopathy (HCM) have been historically noted to have a significantly worse survival over non-syndromic HCM for reasons that are not well understood.12 ,14 ,15 The aims of our study were to describe the spectrum, management and prognosis of heart disease in NS and to investigate the cohort for genotype-phenotype correlations.
Methods
An international Harvard-based phenotype registry and DNA repository for patients with NS was combined with the medical records of all clinically diagnosed NS patients at Boston Children's Hospital (BCH) before 2010. Ethical board approval was sought and provided by the BCH Institutional Review Board. Inclusion criteria required that a patient had a clinical diagnosis of NS and had at least one evaluation by a cardiologist including an echocardiogram or cardiac catheterisation delineating cardiac anatomy. Patients with a concomitant genetic diagnosis in addition to NS were excluded from the study. The data were retrospectively reviewed for descriptors of cardiac morphology, clinical course and for pathogenic mutations in the RAS-MAPK signalling pathway.
Genetic analysis
Patients who underwent genetic screening had DNA amplification and sequencing (multiplex ligation-dependent probe amplification or equivalent) from peripheral lymphocytes in one or more of the eight described genes, namely PTPN11, SOS1, RAF1, KRAS, BRAF, SHOC2, NRAS and CBL.2 Deletions or duplications of one or more exons of these genes were not screened for in this analysis.
Data analysis
For patients who underwent balloon dilation of the pulmonary valve, baseline characteristics were compared for those who required subsequent surgical or catheter intervention versus those who did not, using Fisher's exact test for categorical variables and either the unpaired t test or Wilcoxon rank sum test for continuous variables. Patients who underwent genetic mutation analysis for PTPN11, SOS1, RAF1, SHOC2 or KRAS were compared by cardiac phenotype for presence or absence of any mutation within the described gene using Fisher's exact test. A predetermined p value of 0.05 was set as the threshold of statistical significance. Kaplan-Meier survival probability estimates were generated for those patients with HCM.
Results
We identified 293 patients with NS that met inclusion criteria: 133 patients from the Harvard-based phenotype registry, 114 patients from the records of BCH and an additional 46 patients present in both databases that were included only once. Genetic screening for a NS mutation had been performed in all Harvard registry patients and 50% of patients followed at BCH. The median age of the combined cohort at last follow-up was 14 years; 60% were male. There was no significant difference in the age of those followed at BCH compared to the Harvard registry but a higher prevalence of CHD was noted at BCH (85% and 77%, respectively; p=0.04). Three quarters of the combined cohort were born in the last 25 years (figure 2).

Age range of patients with Noonan syndrome by year of birth.
Cardiac lesions
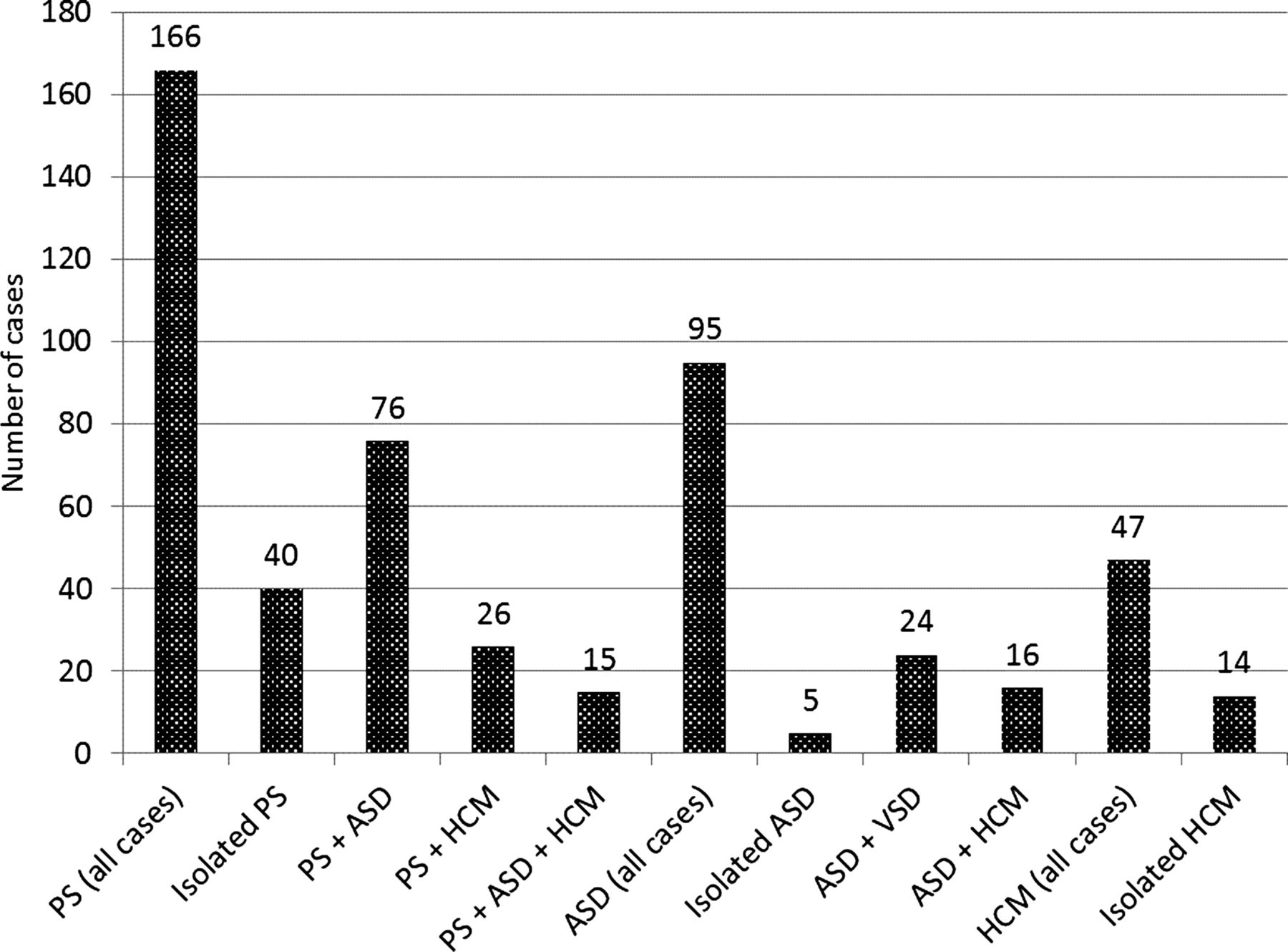
Heart disease was identified in 81% of all patients (see figure 3). It was observed that a high proportion of those NS patients identified with any cardiac defect frequently had more than one isolated cardiac lesion. For example, in those NS patients with valvar PS (n=166), it was observed to be an isolated cardiac finding in 24% (n=40). The majority (76%, n=126) of NS patients with PS, however, had additional concomitant cardiac lesion(s), commonly including secundum atrial septal defects (ASDs) (n=76), HCM (n=26) or both (n=15) (see figure 4).

Congenital heart disease in Noonan syndrome (n=293). Rarely reported cardiac findings included persistence of left superior vena cava to coronary sinus (n=5), complete atrioventricular canal defects (n=3), congenital mitral stenosis (n=3), primum atrial septal defects (n=2), double aortic arch (n=2), anomalous pulmonary venous return (n=2), sinus venosus defect (n=2), bilateral superior vena cavae (n=1) and quadricuspid aortic valve (n=1).

Concomitant cardiac lesions associated with pulmonary stenosis and hypertrophic cardiomyopathy in patients with Noonan syndrome. PS, pulmonary stenosis; ASD, atrial septal defect; HCM, hypertrophic cardiomyopathy.
Valvar pulmonary stenosis
The most common congenital cardiac lesion was valvar PS (n=166, 57% of all patients). Management of valvar PS is summarised in figure 5. Surgery or PBPV was performed in 47% (n=78) of all patients with valvar PS. Of those undergoing PBPV (n=43), the median age at intervention was 6.4 months. The average initial peak pulmonary valve gradient was 65 mm Hg prior to PBPV with an average residual valvar PS gradient of 40 mm Hg. When specifically described, pulmonary valve morphology was noted as dysplastic in 86% of those patients undergoing PBPV. Of those patients who underwent PBPV as their first intervention, 65% (n=28) required further intervention (either repeat PBPV or surgery) for clinically significant residual valvar PS. The only statistically significant risk factor for re-intervention in those with valvar PS was younger median age at the time of initial PBPV (0.54 vs 0.98 years, p=0.046).
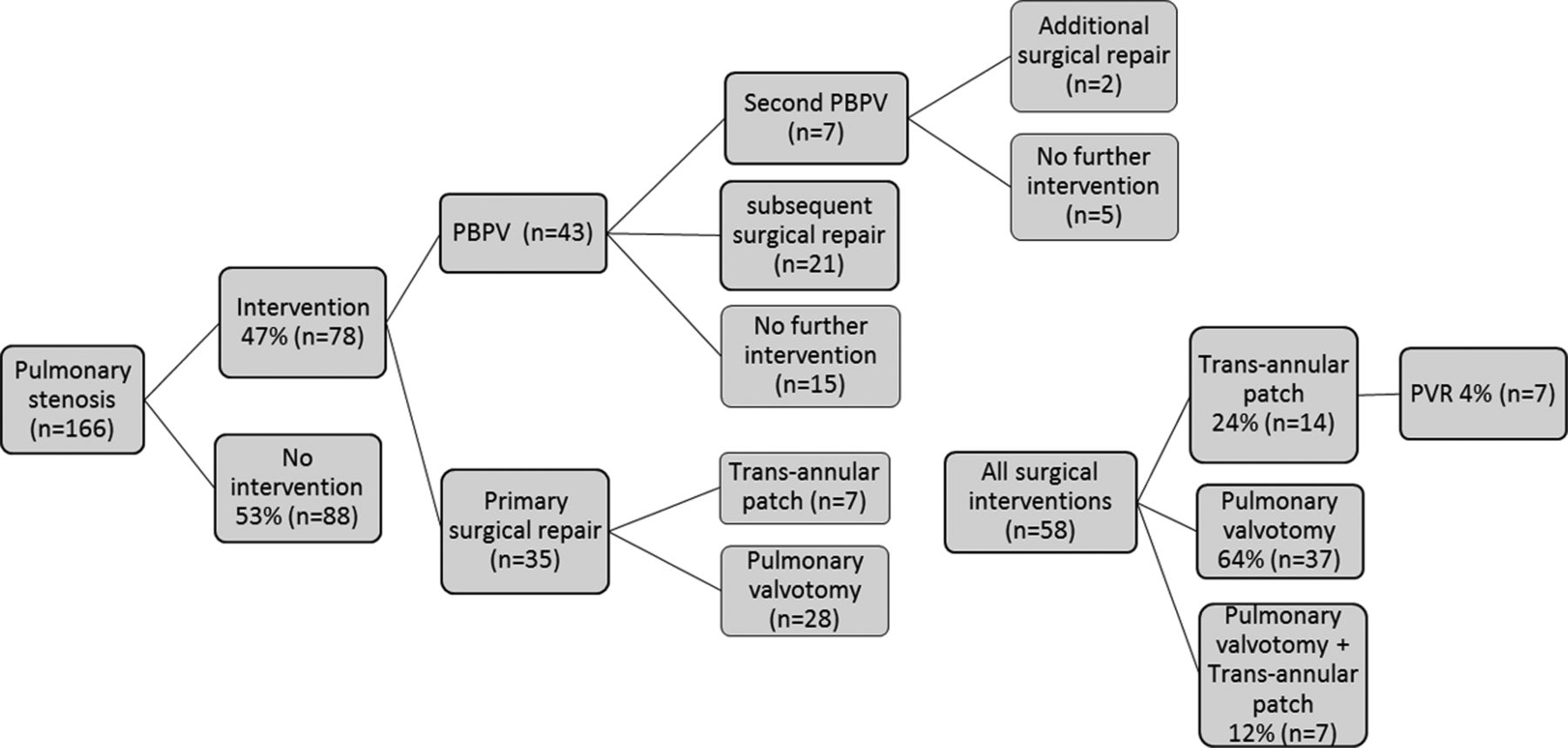
Outcome of patients with Noonan syndrome and pulmonary stenosis. PBPV, percutaneous balloon pulmonary valvuloplasty; TAP, transannular patch; PV, pulmonary valvotomy; PVR, pulmonary valve replacement.
Primary surgical repair for valvar PS was performed in 35 patients with either a pulmonary valvotomy (n=28) or transannular patch (n=7), at an average age of 6.5 years. As a group, patients who had valvar PS managed with primary surgical repair reflected a cohort 17 years prior to that undergoing primary PBPV, likely an era effect in management strategy. To date, 4% of all patients (n=7) with PS have required pulmonary valve replacement at a median age of 30 years.
Hypertrophic cardiomyopathy
There were 47 NS patients diagnosed with HCM with an average age of 16.7 years (range 0.9–59.7 years) by close of study. A diagnosis of HCM was made in the first year of life in 57% (n=27). The degree of wall thickness was described as ‘severe’ echocardiographically in 38% (n=18) with complicating subaortic obstruction noted in 15% (n=7). Concomitant cardiac lesions were found in 70% (n=33) of all patients with a diagnosis of HCM (see table 1). A RAS-MAPK signalling pathway mutation was identified in 44% (n=21) of patients (table 1).
Hypertrophic cardiomyopathy in Noonan syndrome (n=47) by gene mutation
Of all patients with HCM, a subset (17%; n=8) diagnosed in infancy had regression of ventricular wall thickness over time on serial echocardiography with normalisation of ventricular wall dimensions over an average of 3 years (range 1–8 years) and no recurrence noted at an average of 7-year follow-up. Spontaneous regression of HCM was seen in patients with mutations in PTPN11 (n=3), RAF1 (n=1), SOS1 (n=1) and BRAF (n=1).
Interventions for symptomatic HCM (defined as congestive heart failure or exercise-induced systemic hypotension) included extensive myectomy (n=4) and alcohol septal ablation of the first septal perforator (n=1).
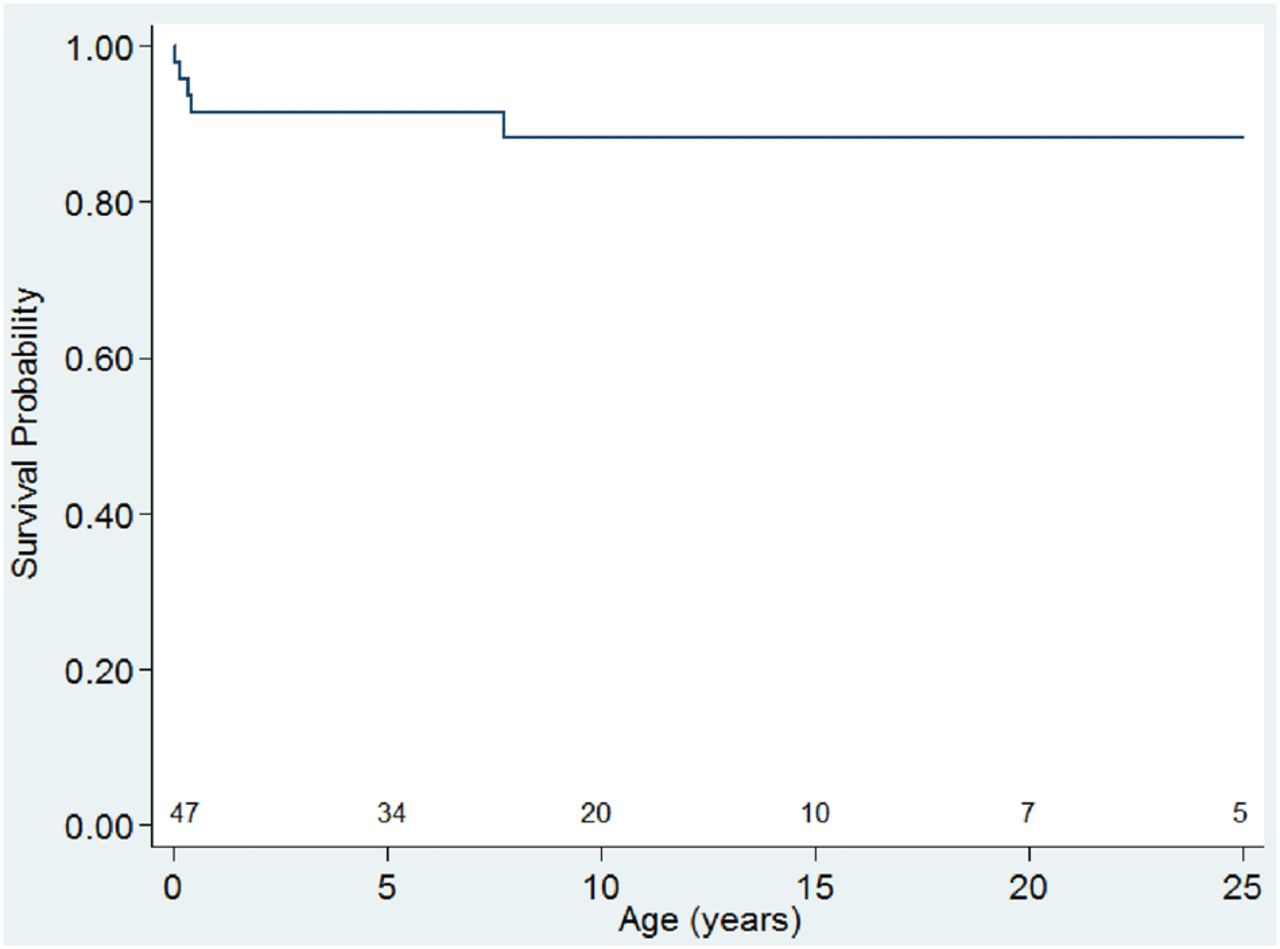
Documented arrhythmias complicating HCM included supraventricular tachycardia in five patients. Causes of death were known for six of the seven HCM patients who died. Refractory heart failure was the primary aetiology contributing to death in five patients with a further sixth patient dying from complications related to prematurity. Of the seven patients with HCM who died, four were diagnosed with HCM in the first year of life, giving this cohort under 1 year of age (n=27) an overall 15% mortality. See figure 6 for a Kaplan–Meier estimation of survival with a diagnosis of HCM.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Kaplan–Meier survival curve for patients with Noonan syndrome and hypertrophic cardiomyopathy up to 25 years of age. Numbers still at risk are shown at 5-year age intervals.
RAS-MAPK signalling pathway mutations
A RAS-MAPK signalling pathway mutation was screened for in 75% (n=219) of patients with identification of a pathogenic gene mutation in 62% (n=136) (see table 2). Among all NS patients screened, mutations in the gene PTPN11 were most frequently reported (41%, n=89). The subgroup of NS patients harbouring a PTPN11 mutation were at significantly higher risk of carrying a clinical diagnosis of either valvar PS (p<0.001) or secundum ASD (p=0.001). SOS1 mutations were identified in 11% (n=25). RAF1 mutations were identified in 3% (n=7) with HCM seen in six patients, making this a significant association (p<0.001). The remaining RAS-MAPK signalling pathway pathogenic mutations identified included BRAF (n=8; 4%), SHOC2 (n=3; 1%) and KRAS (n=3; 1%).
RAS-MAPK signalling pathway mutation and incidence of cardiac defect(s)
Lymphedema
We investigated the incidence of postoperative pericardial and pleural effusions in the cohort of NS patients who underwent surgical repair of either valvar PS or an ASD at BCH (n=119 operations). Pericardial effusions occurred postoperatively in 5.9% (n=7). In the same cohort, 5% of NS patients (n=6) developed clinically significant postoperative pleural effusions with one patient developing a pleural and pericardial effusion simultaneously.
Discussion
This clearly defined NS cohort provides comprehensive insight into the spectrum of heart disease, cardiac management and prognosis in a large number of patients over a broad range of ages. The cardiac lesions identified are similar in prevalence to that described by others.5 ,10 ,16 ,17 Aortic root dilation, seen in a small minority in our cohort (5%), has been previously reported in isolated cases of NS.18–22 No patient in our cohort with aortic root dilation had vessel dissection or required surgical intervention. Atrioventricular canal defects were described in 2.1% patients. This is a smaller percentage than the 5% reported by others, although absolute numbers of patients in any cohort remain small.5 ,23 With regard to the theoretical potential for a higher incidence of lymphatic complications in patients with NS after cardiac surgery, postoperative rates of pericardial or pleural effusion in NS are no higher than those reported for the same cardiac lesions (valvar PS or ASD) in non-NS patients.24–26
We focused on the management of valvar PS given this was the predominant cardiac lesion in patients with NS and accounted for a significant proportion of the associated cardiac morbidity. McCrindle et al13 have previously described an association between poorer long-term relief from valvar PS after PBPV in patients with NS. Although this did not bear out in subsequent logistic regression analysis, the associated dysplastic morphology of the pulmonary valve leaflets in NS patients was thought to be the root cause of a suboptimal response. The incidence of re-intervention after initial PBPV in our NS cohort (65%) is in marked contrast to 5–10% quoted for re-intervention rates after initial PBPV in unselected cohorts of patients with valvar PS.27 ,28 Patients with NS are recognised to have dysplastic pulmonary valve leaflets that may inherently be more refractive to relief of obstruction by PBPV. Despite this high re-intervention rate after PBPV, a significant number of patients with NS and valvar PS (35%) continued to have a therapeutically successful result from PBPV.
Given the theoretical concern for a potential misdiagnosis of NS among the older patients in our database biasing our major conclusions, we performed a subgroup analysis for those with a confirmed RAS-MAPK signalling pathway mutation and valvar PS (n=85). We noted a similarly high incidence of patients requiring intervention for their PS (38%) and of those requiring re-intervention after PBPV (46%). It is worth emphasising that, although only 4% of our total NS cohort with valvar PS have had valve replacement to date at a median age of 30, this may be a relative underestimate, given the median age of our cohort was 14 years of age.
We report distinct characteristics of HCM in a NS cohort; notably, a high incidence of concomitant CHD (70%), early age of presentation (57% in the first year of life) and a 15% mortality in those diagnosed with HCM in infancy. The incidence of a concomitant congenital heart defect in non-syndromic HCM is relatively infrequent at approximately 2.5% of all patients, setting NS HCM distinctly apart.12 Idiopathic HCM presented in the first year of life in 36% of cases in a large patient series.29 The observed higher rate of NS HCM presentation in infancy (57%) may be partly due to empiric screening of patients with NS for CHD once a diagnosis of NS has been made, thus pre-empting symptomatic HCM in some.
Mortality in infants under a year of age in our cohort is significantly less than that reported by others.15 Taking into account that one of the four infant deaths in our series was as a complication of prematurity (27 weeks gestation) only three deaths were directly attributable to HCM (11%).
Spontaneous regression of HCM was seen in 17% of patients (n=8). Two infant patients had severely symptomatic HCM on echocardiography that entirely resolved. Such dramatic regression has, to the best of our knowledge, not been previously reported in NS. Whether a maturational change in the myocardial contractile state is contributing to the regression or an alteration in the RAS/MAPK signalling pathway over the first year of life is not clear at this time. Finally, two patients with PTPN11 mutations and clinically severe HCM had concomitant pathogenic mutations identified in the sarcomeric gene MYBPC3. They were not included in our results or data set (concomitant genetic diagnoses being an exclusion criterion) but warrant comment in considering other contributing genetic factors in NS patients with severe or symptomatic HCM.
We noted a relatively low incidence of ventricular arrhythmia (present in one patient after alcohol septal ablation) in our NS HCM cohort compared to an incidence of 19.6% in those with idiopathic or familial HCM.30 Our NS subjects are relatively young with an average age of 16.7 years and longer term follow-up is required to adequately elucidate the burden of arrhythmias later in adulthood.
Finally, PTPN11 mutations were significantly associated with valvar PS and secundum ASD, observations previously reported.5 ,31–34 Others have also described our finding of RAF1 mutations being associated with HCM.7 ,35–37 Given the significant implication of a diagnosis of NS to an individual patient, it is prudent to consider this diagnosis in the differential for children presenting with CHD and any other phenotype consistent with the presentation. Careful family history and referral for genetic counselling facilitates this investigation.
There are some important limitations of our study. We presume a referral bias to BCH for NS patients with CHD. The findings in our data set may not extrapolate to individuals diagnosed with NS secondary to cascade screening of first-degree family relatives after identification of an index case. It is reasonable to assume that such identified individuals may have a milder phenotype than those who present with clinical features diagnostic of NS or with the associated clinically relevant heart disease. The disparate ages of the cohort must be factored into any interpretation of management strategies and outcomes over time. Patients who underwent genetic screening for a NS mutation were not uniformly re-screened for mutations in newly reported genes as they became clinically available.
In conclusion, we have comprehensively described the burden of cardiovascular disease in a large, clearly defined cohort of patients with NS. Focusing on the management of those with valvar PS, we noted a high re-intervention rate after PBPV. In those patients with HCM, we noted an early age of diagnosis, a high incidence of concomitant CHD and the possibility of spontaneous disease regression in a minority over time. With a deeper understanding of disease characteristics distinct to NS, clinicians will be better able to counsel families regarding prognosis and tailor therapy for optimal patient outcome.
Acknowledgments
We thank Ellen McCusty & Judith Geva for assistance with database management. We would like to thank Emily Harris for assistance formatting figures. We are indebted to the dedicated patients and families with Noonan syndrome who volunteered for the Harvard Noonan syndrome research study, W. Robinson (The Noonan Syndrome Support Group) and the referring physicians.
References
Footnotes
-
Contributors TWP designed the study, assembled the data and wrote the drafts and revisions of the manuscript prior to submission. He is guarantor. KG provided statistical analysis for the data. ETD assisted with extracting data from the Harvard-based phenotype registry. LP assisted with data collection as it related to the cardiac intensive care unit management of Boston Children’s Hospital patients. RSK was the clinical lead on the Harvard-based phenotype registry and DNA repository. AER oversaw and collated the Harvard-based phenotype registry and DNA repository. She provided assistance with the concept, design and subsequent review prior to submission for publication. RVL, as the senior author, provided assistance in the concept, design and subsequent review prior to submission for publication.
-
Funding NIH T32 GM007748 Grant: Genotype Phenotype Correlations in Noonan Syndrome and Related Disorders (A.E.R.), NIH K12 Grant HD052896-01A1: The Child Health Research Center; Genotype Phenotype Correlations and Gene Discovery for the Disorders of the RAS-MAP Kinase Pathway (A.E.R.)
-
Competing interests None.
-
Ethics approval Boston Children’s Hospital IRB.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Study data available to other investigators pending Institutional Review Board approval at Boston Children’s Hospital. Please contact corresponding author for further inquiries.