Article Text

Abstract
1. Microcephaly is a clinical finding, not a ‘disease’, and is a crude but trusted assessment of intracranial brain volume.
2. Developmental processes reducing in utero neuron generation present at birth with ‘Primary microcephaly’.
3. ‘Secondary microcephaly’ develops after birth and predominantly reflects dendritic or white matter diseases.
4. Microcephalic conditions have a heterogeneous aetiology, but increasingly genomic tests are available that allow an exact diagnosis.
- Paediatric Practice
- Neurodevelopment
- Neurology
- Genetics
Statistics from Altmetric.com
Introduction
This article provides a diagnostic structure to follow when the clinician is presented with a child with microcephaly. The brain in these children either didn't grow to the normal size during pregnancy (primary microcephaly) or had reduced growth later (secondary microcephaly). They may have suffered an injury, or have a genetic disorder affecting brain function and postnatal growth. In our clinical practice, we find it useful to consider primary and secondary microcephaly separately, and present a diagnostic algorithm. History and examination are essential in guiding towards the correct investigation and diagnosis. Head circumference charts are numerous—and different reports have used different charts—but in practice this is rarely a clinical problem. Over the last two decades a legion of genetic disorders have been reported, many of which include microcephaly as a significant feature. The genetic aetiology of these disorders is being progressively defined and is already known for many. Current sophistication in radiological and genetic investigations allow for their ready diagnosis. Despite this, considerable work still needs to be done to determine the cognitive/behavioural profiles and natural history of these recently emerged disorders.
Relevant good reviews are of, an historical overview of microcephaly,1 microcephalic syndromes,2 investigations for microcephaly,3 the genetics of microcephaly4 and a radiological classification of cerebral malformations and developmental anomalies.5 We have tried to select further references with the clinician in mind, and include table 2 giving information sources for diagnoses of microcephaly with dysmorphic features and/or congenital anomalies and similarly table 3 giving information sources for microcephaly caused by chromosome perturbations.
Microcephaly: nomenclature matters
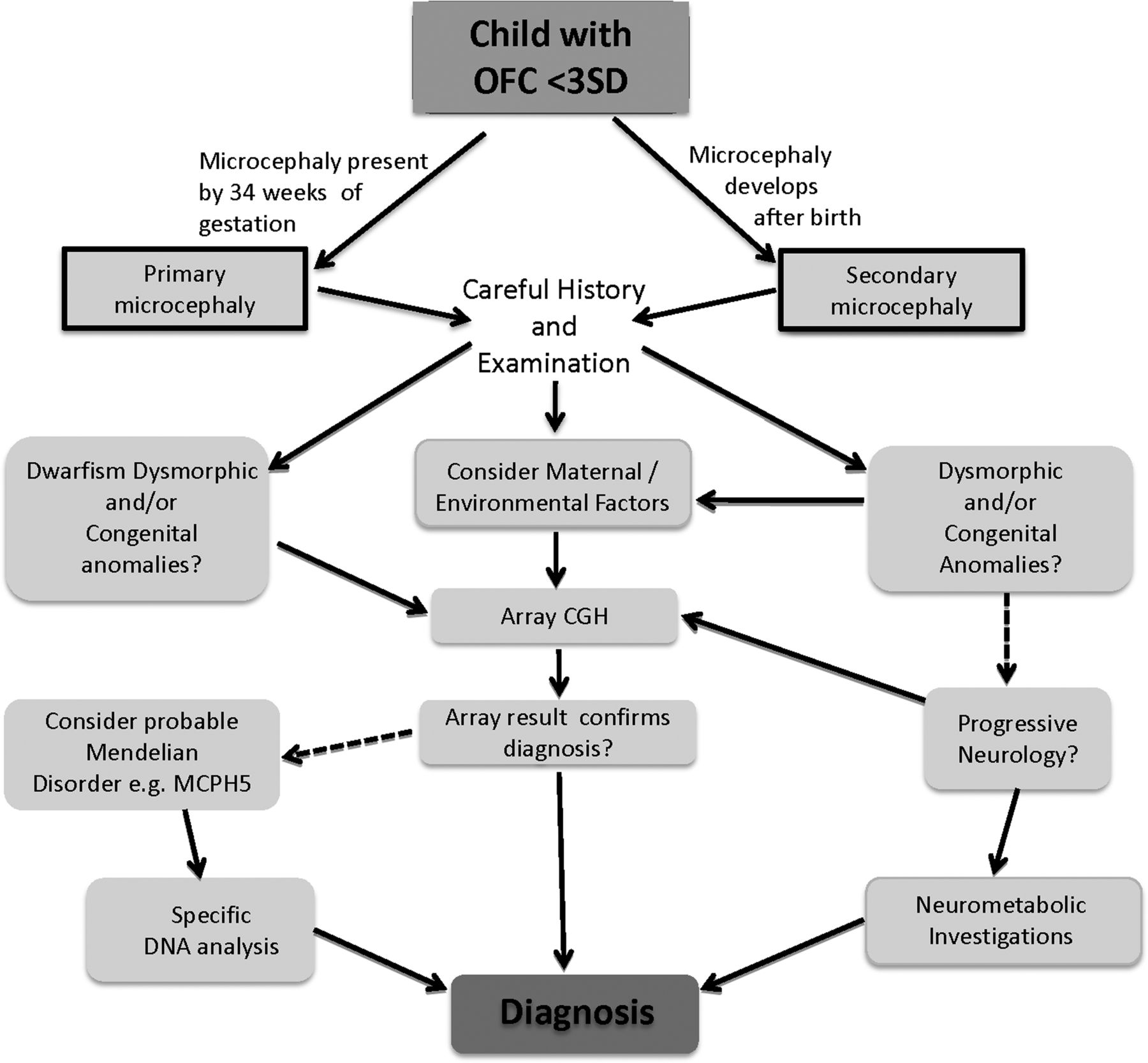
It is important to be clear that the term ‘microcephaly’ is a clinical finding, and should not be used as a disease designation. It is commonly defined as a significant reduction in the occipital-frontal head circumference (OFC) compared with age and sex (but sometimes not ethnically) matched controls.1 Arguments have been made for an OFC of <−2 SD or <−3 SD to be used to define a person less than 19 years old as having microcephaly. We use an OFC of >3 SD below the age and sex expected mean before use of the diagnostic algorithm (see figure 1). Our justification for this is driven by:
-
Using the label of microcephaly in the −2 SD to −3 SD population, does not help with management as a significant proportion of these children will be ‘normal’;
-
Yield from genetic/investigation is low;
-
Head shape matters as a round head contains a greater volume of brain than an elliptical one of the same circumference.
{kind=link}
Clinical diagnostic algorithm for microcephaly. Full arrows are a ‘Yes’ decision, and broken arrows for a ‘No’ decision.
The clinical approach should be organised by history, examination, then thinking, and finally investigation. First, determine if the small head size was present at birth (primary microcephaly) or developed postnatally (secondary microcephaly). Usually an easy task, but if there were no birth head circumference measurement or there is doubt that the measurement was accurate this can be difficult—and in such cases serial measurements are useful; a reduced occipitofrontal head circumference (OFC) that follows a growth curve parallel to normal would usually suggest a primary microcephaly, whereas an OFC that falls relative to the normal growth curve is more typical of a secondary microcephaly. Thereafter, we divide cases into those: ▸ In which there is an environmental aetiology, for example, a spastic diplegia, and brain imaging showing periventricular leucomalacia typical of intrauterine or birth asphyxia; ▸ In which there are dysmorphic findings and/or congenital malformations, for example, small size, distinctive facial appearance and ulnar ray anomalies found in the de Lange syndrome; ▸ In which there are other distinctive clinical features, for example, secondary microcephaly, severe developmental delay, episodic hyperventilation and the hand-wringing seen in Rett syndrome. Progressing along each of these phenotypic avenues towards a correct diagnosis may then require a confirmatory test or more wide-ranging investigations and possibly referrals to other specialists. Such referral would be to a paediatric neurologist if there is progressive neurology or a neurological/neurometabolic diagnosis and to a clinical geneticist if there are potentially pathogenic array or gene mutation results or if a diagnosis of a dysmorphic syndrome or a Mendelian genetic disorder is suspected.
We note that in the early stage of conditions such as metabolic disorders, the OFC will diverge from its previous centile but will not lie below −3 SD for some time, during which investigation and diagnosis may be needed. So the decelerating OFC needs expert assessment.
Clearly, in a child with a history of asphyxia and an OFC −2 SD to −3 SD beneath the mean, the head circumference is likely to reflect brain injury, and furthermore specific genetic and metabolic investigations have a low yield with consistent tracking in this centile as opposed to decelerating growth.
Different OFC charts exist, and are at their most divergent in the 1st year of life. Sex specific charts must be used as generally men have slightly larger brains; although women have a later closure of the anterior fontanel. (This is a consequence of the greater muscle mass of men compared with women). It is important to use the same chart for all measurements for a child, and one that is as ethnically suitable as possible, for example, for children living in Britain the UK-WHO growth charts.6
Within the normal range of head circumference, <+3 SD to >−3SD, there is little correlation with IQ. However, outside of these limits the greater the deviation the greater the likelihood and severity of learning difficulties (there are a very few exceptions, eg, Bloom’s syndrome, see later). As the degree of deviation from normal can be difficult to quantitate from standard charts we use an OFC SD table to guide us, see table 1. For children with more significant microcephaly, <−8SD it can be more difficult to measure OFC (as the occiput may be sloping and the child's hair can make an undue contribution) but attention to the landmarks of forehead, just above both ears and symmetry of positioning will ensure a correct measurement.
Occipitofrontal head circumference (OFC) SD for men and women up to the age of 18 years
Primary microcephaly
Primary microcephaly is defined as microcephaly being detectable prior to 36 weeks of gestation. Thus the primary aetiology could be: a failure or reduction in neurogenesis of neurons and possibly other white matter elements (eg, first trimester cytomegalo virus (CMV) infection, chromosomal disorder or autosomal recessive primary microcephaly); a destructive event prior to birth (eg, prenatal formation of porencephalic cysts due to hypoxia/ischaemia, or some mitochondrial mutations); and much more rarely, a very early onset degenerative process (eg, some cases of Aicardi-Goutieres syndrome). So history is vital to determine if there are prenatal environmental/maternal causes for a case of primary microcephaly, and if suspected the appropriate TOxoplasma/Rubella/Cytomegalovirus/Herpes (TORCH)±HIV screen, or MRI scan can be performed. Scrutiny of maternal notes should confirm the diagnosis: for example, phenylketonuria (rare now, but still possible if an affected mother strays from her strict diet during pregnancy), malnutrition or hypothyroidism; teratogens such as alcohol (noting that foetal alcohol syndrome is a difficult diagnosis to make7), recreational and medicinal drugs; or placental insufficiency. Brain disruptions can be caused by a twin pregnancy, maternal abdominal injury, placental abruption, maternal antibodies, but in some cases the aetiology remains undiscoverable.
The timing of the onset of OFC reduction is crucial, but is usually discernible from prenatal ultrasound scans and birth-growth measurements. If the microcephaly is detected prenatally prior to 20 weeks then a severe outcome is to be expected, as well as a high likelihood of brain malformations. Such cases usually result in extreme primary microcephaly with an OFC of <−8 SD. Foetal brain imaging is warranted to assist with diagnosis and prognosis. However, in most conditions causing primary microcephaly the foetal head circumference is within the normal range at the 18–20 week-ultrasound ‘anomaly scan’.
For those with primary microcephaly and an OFC between −3 SD and −8 SD (see figure 1), we divide into:
-
Primary microcephaly with dwarfism. It is important to distinguish between proportionate growth retardation—where length, weight and OFC are reduced to a similar degree, and disproportionate growth retardation—where the OFC is reduced to a greater extent than length and weight.
In proportionate growth retardation, microcephaly would not be the usual reason for referral; however, a number of disorders should be considered. In microcephalic osteodysplastic primordial dwarfism type 2, prenatal and postnatal growth retardation are severe (at birth average height is −7.0 SD, weight is −3.9 SD and OFC −4.6 SD, and at skeletal maturity, they are −10.3 SD, −14.3 SD and −8.5 SD, respectively8), the child has a Seckel syndrome like face, often hip dysplasia, poor dentition and minor learning difficulties. This phenotype is autosomal recessive and can be caused by mutations in PERICENTRIN and DNA LIGASE4 (where progressive pancytopaenia and lymphoreticular malignancies also occur).9 ,10
The chromosome breakage disorders—Bloom’s syndrome, and more rarely in Fanconi anaemia—can present as proportionate growth retardation unresponsive to adequate nutritional intake. Most importantly, despite significant microcephaly, intelligence in Bloom's syndrome and Fanconi anaemia syndrome is normal.11 ,12 Confirmatory testing is by specialised cytogenetic studies and gene sequencing; RECQL3 for Bloom's syndrome, and >10 genes for Fanconi anaemia syndrome including biallelic mutations in the familial breast/ovary cancer gene BRCA2.
In disproportionate growth retardation, where the degree of microcephaly is more affected than body growth, the child is often classified as having a Seckel syndrome phenotype. Such children almost always have more significant learning problems. Seckel syndrome is usually autosomal recessive and can be caused by mutations in a growing number of genes including ATR, RBBP8, CPAP, CEP152 CEP63, DYRK1A, NIN and ATRIP.13–15
-
Primary microcephaly, but with dysmorphic features and/or congenital anomalies.
In this group a chromosome perturbation must be sought using a ‘microarray’, and consideration given to referral to clinical genetics.16 There are a large and growing number of chromosome perturbations causing microcephaly, learning difficulties and (intrafamilial and interfamilial) variable additional features. Only a minority are recognisable by phenotype alone, such as 4p- (the Wolf-Hirschhorn syndrome), and local cytogenetic and clinical genetics services are important to determine whether the genotype/phenotype are in accord, as well as interpreting online databases such as DECIPHER.17 In general, the greater the number of genes involved, the more severe the phenotype. With the increasing resolution of microarrays smaller deletions and duplications have become detectable (often called InDel—insertions and deletions, or CNV—copy number variants). Many of these small findings (involving a few genes or even a single exon, especially if a duplication) are not pathogenic. Others can ‘contribute’ but not cause a phenotype, and a clinically important phenotype only results if there are other genomic changes—thus array results may be difficult to interpret at present.18
Children with ring chromosomes can present with proportionate growth retardation, learning difficulties and usually ragged café-au-lait and hypomelanotic macules (these may be distributed in the lines of Blashkow). If a ring chromosome is sought then discussion with a cytogenetic laboratory is essential to allow them to decide whether an array or cytogenetic spread with increased number of cells counted would be the better diagnostic approach.
Dysmorphic features and/or congenital anomalies and growth retardation occur in Mendelian disorders. A representative, and not so rare, example is the de Lange syndrome.19 The phenotype has so distinctive a facies with synophrys, symmetrical growth retardation and often ulnar ray anomalies that a clinical diagnosis can often be made and confirmed, if necessary, by gene testing of components of the cohesion complex, including the genes NIPBL, SMC1A, SMC3, RAD21 and HDAC8. An example of a recessive disorder is the Meier-Gorlin syndrome where growth retardation (typically −4 SD for all parameters) is combined with bilateral microtia (97%), aplasia or hypoplasia of the patellae (94%), in women mammary hypoplasia (100%) and normal development.20 Mutations are in the prereplication complex genes, which assemble as a protein complex prior to DNA replication (including the genes ORC1, ORC4, ORC6, CDT1 and CDC6).21
-
Primary microcephaly with additional clinical features.
In this group the additional features are usually predominant such as in mitochondrial and metabolic disorders, which we do not consider further, but see tables 2 and 3 and ref. 3.
Information sources for microcephaly with dysmorphic features and/or congenital anomalies
Information sources for microcephaly caused by chromosome perturbations
A number of ‘disorders of cerebral development’ present with primary microcephaly and the following should lead to early consideration of a brain scan allowing their diagnosis; extreme microcephaly (OFC <−8 SD), early onset of epileptic seizures especially if difficult to control, additional central nervous system (CNS) signs such as spasticity, excess drooling, unilateral motor signs and severe/profound learning difficulties. A growing number of gene mutations have been reported that cause ‘disorders of cerebral development’. However, the range of phenotypes that can be caused by different types of mutations in each gene is currently unclear and continues to expand. Examples are the X linked disorder ATRX (which can also be associated with genital hypoplasia), the recessive disorders caused by biallelic mutations in WDR62 (non-sense mutations are associated with almost any cerebral anomaly, and sometimes marked cerebral asymmetry) and NDE1 (causing extreme microcephaly and profound developmental delay) and new dominant mutations in TUBA1A (associate with lissencephaly and spasticity) and TUBB2B (causing asymmetric polymicrogyria).22 ,23
The rare DNA repair disorders can be difficult to diagnose initially, such as: Nijmegen breakage syndrome in a child with immune deficiency, primary microcephaly and mild learning difficulties; and trichothiodystrophy in the sun-sensitive child with brittle sparse hair.23
-
‘Autosomal recessive primary microcephaly’ phenotype.
This phenotype is a diagnosis of exclusion in a child with primary microcephaly, developmental delay of mild–moderate degree with a normal brain scan (note common finding of wide cerebro-spinal fluid (CSF) spaces) but no other abnormal growth, and dysmorphic or neurological features. Other potential causes such as prenatal insult, chromosome perturbation and dominant microcephaly must be eliminated. The disorder is recessive and caused most commonly by mutations in ASPM (non-sense mutations) and WDR62 (mis-sense mutations).24
Secondary microcephaly
Secondary microcephaly is defined as microcephaly developing after birth. Thus the primary aetiology is not a failure of neurogenesis. The cause is anything that disrupts the orderly development and functioning of the central nervous system! This includes neural migration disorder of neurons, a prenatal insult, a block to normal development and a degenerative process. So, secondary microcephaly will rarely be the primary presenting feature. History and examination are again vital to define the phenotype and its progression particularly seeking significant prenatal, birth and environmental events, as discussed above in the primary microcephaly section.
The most important factor in our approach to secondary microcephaly is whether the child has a static or progressive disorder (see figure 1). If static, and if the phenotype includes dysmorphic features and/or congenital anomalies, then again the two major diagnostic categories are chromosome perturbations and Mendelian single gene disorders. The majority of reported chromosome disorders are associated with developmental delay and secondary microcephaly occurs in at least half of these (the general rules for chromosome perturbations are: deletions are much more severe than duplications; the greater the number of genes involved the worse the phenotype; severe phenotypes get described more frequently than minor ones; and some microarray anomalies simply cannot be currently classified). A good example of a secondary microcephaly microdeletion is Miller-Dieker syndrome caused by a deletion of chromosome 17 p13.3. The larger deletions are associated with a more severe phenotype of lissencephaly/pachygyria with profound intellectual handicap and often a distinctive facies. Smaller deletions only involving the LIS1 gene (and point mutations in LIS1) cause a less severe form of lissencephaly/pachygyria.25 Rubinstein-Taybi syndrome is a good example of one of the very many Mendelian disorders causing secondary microcephaly and learning difficulties. Diagnosis is aided by distinctive facies, broad thumbs/big toes and postnatal growth retardation. Diagnosis is clinical and can be confirmed with finding mutations in CREBBP, EP300 or SRCAP.26
If the secondary microcephaly is associated with progressive neurological findings, then first metabolic diseases should be considered but are a rare cause of secondary microcephaly, about 1%.3 Usually progressive neurological changes precede the secondary microcephaly resulting in investigations for neurometabolic diseases being first performed. A large number of genetic disorders can cause this phenotype ranging from the ill-understood disorders of cortical function such as Rett syndrome (with its halt in development about the first year and distinctive stereotypic hand movements) and PEHO syndrome (with unusual oedema of the back of the hands/feet and optic atrophy), through DNA repair disorders such as ataxia-telangiectasia and Cockayne's/MICRO syndromes (where additional features such as the bulbar telangiectasia or progressive deafness and lipoatrophy help with diagnosis) to dysmorphic conditions such as Cohen syndrome (with distinctive snarling facies, and obesity, and then blindness developing in the second decade).27 It is always important to examine parents as autosomal dominant microcephaly is best diagnosed by a family history, and can be associated with leg lymphoedema and choroidoretinal lacunae (usually completely symptomless).
Confirmatory diagnosis by DNA testing is available for the majority of these Mendelian causes of secondary microcephaly, and with the real advances in next generation DNA sequencing it seems likely gene sequencing will soon be the predominant confirmatory test we perform. This process will be hastened in the near future by the developments of panels of genes grouped by phenotype, rather than sequencing each gene separately.
Comments on investigations
-
Microarray testing is an almost obligatory test for a child referred with microcephaly. Microarrays are manufactured to offer differing resolution, but the greater the resolution the greater the number of anomalies found, but beware as most of these are either harmless polymorphisms and not clinically relevant or cannot currently be classified. For advice, see table 3.
-
Brain scans: Not every child needs brain imaging, especially as sedation or a general anaesthetic will be needed in many cases. However, it should be considered in any child with primary microcephaly with an OFC<−6 SD, or significant learning difficulties, or early onset epileptic seizures, or motor features (especially spasticity or if they are asymmetric) or if there is a known diagnosis, say autosomal dominant holoprosencephaly, and quantification of cerebral architecture would guide prognosis. The same general rules apply for secondary microcephaly, but with the greatest emphasis on the degree and timing of developmental delay. In children with an OFC 3–6 SD beneath the mean, and no additional features, there is a very low yield, and consideration for other testing more logical.
-
Biochemical and other tests which are diagnostic will be performed as second line targeted testing, for example, 7-dehydrocholesterol for Smith-Lemli-Opitz syndrome, and a MRI/computerised axial tomography (CAT) scan revealing the intracerebral calcification pattern of Aicardi-Goutieres syndrome. In many cases subsequent DNA testing of the causative gene(s) will be undertaken, but in the near future initial DNA testing may become a quicker, cheaper and more comprehensive approach. We always consider, and have a very low threshold to test, the plasma amino acids, lactate, ammonia, thyroid function, and urine amino/organic acids, purine and creatine guanidinoacetate profiles. It is also important to consider GLUT1 deficiency as a treatable cause of microcephaly via CSF glucose, and if analysing we check CSF lactate to avoid repeat sampling.
-
Gene testing is in transition. Two factors are operating, first the technology to analyse large amounts of DNA quickly, accurately and more cheaply is entering National Health Service (NHS) diagnostics, and second the remaining genes causing Mendelian disorders are being rapidly discovered. This means that for a given microcephalic phenotype all (or almost all) genes that can cause that phenotype can be simultaneously sequenced and the pathogenic mutations sought, increasing the diagnostic yield, and significantly reducing the cost and time taken. Regional genetics services will be able to give advice on current testing available in the UK during this time of rapid change.
Conclusions
The investigation of the child with microcephaly should be driven by a working clinical diagnosis, or diagnostic category, derived from the history and examination. Non-genetic causes of microcephaly have to be sought and where possible proven. Cytogenetic testing will have the highest diagnostic yield. Brain scanning gives prognostic and diagnostic data, the scan being abnormal in >50% cases (using the above criteria for scanning). Increasingly the large number of Mendelian disorders affecting brain size, growth and function are being discovered and phenotype/genotype correlations delineated. This comes at the same time as a revolution in the scope of DNA testing, allowing for a far greater number of specific diagnostic tests, and even the possibility that DNA testing will soon be the primary test performed.
References
Footnotes
-
Contributors CGW and AP were individually responsible for drafting sections of the article, including the tables and the figure; both revised the article and both gave final approval of the article to be submitted for publication.
-
Funding NHS.
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.