Article Text

Abstract
Objectives Intranasal nalbuphine could be a safe, efficacious and non-invasive alternative to parenteral pain medication in infants. We aimed to assess pharmacokinetics (PK) and tolerability of intranasal and intravenous nalbuphine administration in infants.
Methods Prospective open-label study including infants 1–3 months of age admitted to the emergency department, receiving nalbuphine for procedural pain management. Patients were alternately allocated to a single nalbuphine dose of 0.05 mg/kg intravenously or 0.1 mg/kg intranasally. Nalbuphine PK samples were collected 15, 30 and 120–180 min after dosing. Area under the concentration time curve (AUC0-Tlast) was calculated by non-compartmental analysis (NCA) and compared by Wilcoxon test. Neonatal Infant Pain Score was assessed during nalbuphine administration and the following interventions: venous access, urinary catheterisation, lumbar puncture.
Results Out of 52 study subjects receiving nalbuphine, 31 were eligible for NCA (11 intravenous, 20 intranasal). Median AUC0-Tlast after 0.05 mg/kg intravenously was 8.7 (IQR: 8.0–18.6) µg×L/hour vs 7.6 (5.4–10.4) µg×L/hour after intranasal administration of 0.1 mg/kg (p=0.091). Maximum serum concentration (Cmax) was observed 30 min after intranasal administration (3.5–5.6 µg/L). During intravenous and intranasal nalbuphine administration, mild to no pain was recorded in 71% and 67% of study subjects, respectively.
Conclusion This is the first study investigating intranasal administration of nalbuphine in infants suggesting an intranasal bioavailability close to 50%. Non-invasive intranasal application was well tolerated. Additional studies are warranted to optimise dosing and timing of interventions as Cmax is delayed by half an hour after intranasal administration.
Trial registration number NCT03059511.
- pain
- paediatric emergency medicine
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
What is already known on this topic
Nalbuphine is frequently used in infants and neonates but it is only licensed for parenteral use.
Intranasal nalbuphine delivery could be a safe, efficacious and non-invasive alternative to parenteral use in children, especially for infants in the acute care setting.
There are few publications describing intranasal nalbuphine administration, but pharmacokinetic data for the intranasal route of administration are completely lacking.
What this study adds
This is the first study reporting clinical pharmacokinetics of intranasal nalbuphine in infants 1–3 months of age.
We observed similar exposure coverage following single administration of 0.1 mg/kg intranasal and 0.05 mg/kg intravenous nalbuphine, suggesting an intranasal bioavailability of 50%.
Intranasal administration was safe and well tolerated in 67% of patients.
How this study might affect research, practice or policy
This study provides first evidence for feasibility and safety of intranasal nalbuphine administration as an alternative to parenteral use in infants 1–3 months.
Intranasal nalbuphine may offer an additional non-invasive option for pain treatment in infants.
Additional studies focusing on optimal dosing and timing of painful interventions following intranasal administration can be designed based on results of this study.
Introduction
Nalbuphine, an opioid analgesic agent, is often used to treat moderate to severe pain in children.1–4 In the USA, it is licensed for adults only; in the UK, it is not licensed and in some European countries for children aged over 1.5 years at a dose of 0.1–0.2 mg/kg (maximal 10 mg) intramuscularly, intravenously or subcutaneously every 3–6 hours.1 5 6 However, nalbuphine is frequently used in infants and neonates as it shows good analgesia and a lower ceiling effect on respiratory depression in comparison with other opioid analgesic agents due to its unique pharmacological properties as a μ-receptor antagonist/κ-receptor agonist.1–4 7
Intranasal nalbuphine delivery could be a safe, efficacious and non-invasive alternative to parenteral use in children, especially for infants in the acute care setting who are not eligible for fentanyl. The lowest body weight limit for intranasal fentanyl use is 10 kg, due to the supposed risk of potentially severe adverse drug reactions such as apnoea.8–10 Main advantages of intranasal drug delivery are ease of administration, high patient and provider satisfaction, expected rapid onset of action, the avoidance of gastrointestinal and hepatic first-pass effects and ease of use in patients with nausea and vomiting.11–13 Although nalbuphine has been approved more than 20 years ago, pharmacokinetic (PK) data on the drug remain limited, especially in children.2–4 12 14–16 The drug is mainly hepatically metabolised involving several phase I and phase II pathways.17 As a drug with high hepatic extraction, substantial and variable first-pass metabolism limits its oral use.1 3 15 Half-life in adults is 2.5–3 hours1 and similar in children.3 7 There are few publications describing intranasal nalbuphine administration, but PK data for the intranasal route of administration are completely lacking.18 19
The primary objective of this study was to assess PK of nalbuphine in infants 1–3 months of age after single intravenous (0.05 mg/kg) and intranasal (0.1 mg/kg) application, respectively. The used intravenous dosing differs from the above-discussed licensed dosing because the Swiss medical authority did not approve to study 0.1 mg/kg intravenously and 0.2 mg/kg intranasally in infants <3 months due to safety concerns.
The secondary objective of the study was to assess tolerability of intranasal application, pain control and safety after single intravenous (0.05 mg/kg) and intranasal (0.1 mg/kg) administration.
Methods
Trial design and participants
We performed a prospective, single centre, open-label PK study, conducted in the interdisciplinary emergency department of the University Children’s Hospital Zurich between 2017 and 2018. Infants aged 29 days–3 months with minimum body weight of 3.0 kg and fever without a source requiring a partial or full sepsis work-up according to our clinical practice guideline were eligible.20 In febrile infants older than 3 months, the decision of diagnostic and painful procedures is based on the clinical condition of the patient and may show a big variety. Therefore, to ease inclusion criteria, we decided to include infants up to 3 months undergoing the same diagnostic and interventional procedures according to our clinical practice guideline. Main exclusion criteria were premature birth, kidney or liver disease, chronic illness, documented previous adverse reaction to nalbuphine, treatment with other central nervous system depressants within 5 days prior to study, and epistaxis or nose trauma (only for the intranasal application).
Interventions
Following informed consent of the parents, included patients were alternately allocated to either 0.05 mg/kg intravenous (bolus) or 0.1 mg/kg intranasal nalbuphine (Nalbuphin OrPha, 20 mg/ 2mL OrPha Swiss, Kuesnacht, Switzerland); alternation could be switched to balance the number of patients in the two groups. Intranasal dose was doubled compared with intravenous because we expected nalbuphine intranasal bioavailability between 50% and 80%, according to lipophilicity and molecular weight.21 22 Using a 1 mL syringe, the corresponding volume of 0.02–0.1 mL was withdrawn and administered. This volume is well within the range of 0.025–0.20 mL considered suitable for nasal drug delivery as associated with minimal outflow or gastrointestinal absorption through mucociliary clearance.21 23 24 In the intravenous administration group, painful interventions for medical work-up were performed 5 min before nalbuphine administration (venous access for blood sampling) and 20 and 35 min after drug administration (urinary catheterisation and lumbar puncture). A nasal device (Mucosal Atomization Device 300 Teleflex, USA) was used for intranasal administration. In the intranasal administration group, venous access was performed 5 min after nalbuphine administration, urinary catheterisation after 20 min and lumbar puncture after 30 min successively. Blood samples with a minimum amount of 0.5 mL blood to measure nalbuphine serum concentrations were obtained from placed intravenous access in both groups 15, 30 and 120–180 min after drug administration. The same intravenous access was used to administer nalbuphine intravenously and to collect the blood samples. Tolerability of drug administration was assessed by the Neonatal Infant Pain Score (NIPS)25 during intravenous and intranasal nalbuphine administration. Pain control was also assessed by NIPS during each intervention. As safety endpoints, oxygen saturation, heart rate and, if feasible, blood pressure were recorded at baseline and during nalbuphine administration, venous access, PK sampling, urinary catheterisation and lumbar puncture. Adverse events were recorded at any time.
Serum drug analysis
Nalbuphine serum levels were measured using liquid chromatography coupled to tandem mass spectrometry. The lower limit of quantification was 0.1 µg/L and the upper limit of quantification 2500 µg/L. Intraday and interday assay precision was <8.15% and <5.3%, respectively.
PK analysis
Data were analysed by non-compartmental analysis (NCA). NCA included only patients for whom all three serum concentration measurements were available. Implausible high serum concentrations were defined as >60 µg/L (corresponding to a theoretical distribution volume of <0.83 L/kg, that is, much smaller than a previously reported distribution volume of 3.62±1.77 L/kg in children 1.5–5 years)2 or rising concentrations after intravenous administration, were excluded from the analysis. Those values were suspected to be related to protocol errors like missed line flushing between drug administration and blood withdrawal in the intravenous group, an accidental sample switch or a dose calculation error.
Sample size calculation
Assuming an interindividual variability (coefficient of variation) in the area under the concentration time curve (AUC0-infinity) of 68%–78% in this age group,15 a sample size of 19 patients per group (rounded up to 21 to account for possible drop-out) was deemed necessary to demonstrate a significant mean AUC difference between the groups outside the 80%–125% interval, with a power of at least 80% (t-test for mean difference, 5% significance level).26
Non-compartmental analysis
NCA was conducted using package NonCompart in R (V.3.6.1, R Core Team, 2019); for intranasal data, the initial concentration at time=0 min was set to 0 µg/L. Nalbuphine AUC0-Tlast was calculated according to the linear trapezoidal method with linear interpolation; the last two decreasing concentrations were assumed to reflect the terminal elimination phase. Maximum measured serum concentration (Cmax) and time to maximum concentration for intranasal (tmax) were extracted for each patient. AUC0-infinity and % of AUC0-Tlast/AUC0-infinity were only calculated for the intravenous group, as a terminal elimination phase could not be observed for all intranasal concentration profiles. All variables were summarised by median and IQR, and compared between groups by non-parametric Wilcoxon test (instead of t-test used for sample size calculation above, due to remaining non-normal distribution after log-transformation).
Tolerability of intranasal application and pain control
NIPS were categorised as no/mild (0–2), moderate (3–4) and severe (>4) pain, and were summarised as number (%). Tolerability was evaluated as number (%) with mild NIPS during drug administration. Pain control was evaluated as number (%) of patients with severe NIPS during interventions (establishment of venous access, urinary catheterisation and lumbar puncture).
Safety (adverse events and vital signs)
Vital signs were collected at baseline and during each intervention. Other reported adverse events were categorised as mild or moderate (by the treating physician) or serious in case of death, life-threatening events, associated (prolonged) hospitalisation, disability or permanent damage, or required intervention to prevent permanent impairment or damage. Each adverse event was assessed for causality (unrelated or related to nalbuphine) by the treating physician. Events were considered as unrelated if the event started in no temporal relationship to nalbuphine administration or if the event could be definitely explained by underlying diseases or other conditions.
Results
Data
A total of 54 patients were included in the study. Patient characteristics are given in table 1. In 40 patients (19 intravenous/21 intranasal), all three planned nalbuphine concentration measurements were obtained. According to criteria specified above, concentrations from nine study subjects (eight intravenous/one intranasal) were excluded from the PK analyses: three concentrations because of rising values after intravenous administration and six because of implausible concentrations. The medical record of two patients (one intravenous/one intranasal) revealed an accidental (10-fold) overdose. Therefore, a total of 31 patients were eligible for primary PK analysis (figure 1). A total of 52 patients received nalbuphine (26 intravenous/26 intranasal) and were available for tolerability, pain control and safety analysis.

Flow chart: Implausible serum concentrations were defined as implausible high serum concentrations (>60 µg/L, inclusive expected accidental overdoses) or rising concentrations after intravenous administration.
Characteristics of 31 patients included in non-compartmental analysis (NCA) (left) and of all 52 patients who received nalbuphine (right)
Pharmacokinetics
Serum concentration time profiles are shown in figure 2A,B. Calculated NCA PK parameters are summarised in table 2. No significant difference for AUC0-Tlast could be found between the intranasal and intravenous group (p=0.091), and the median intranasal AUC0-Tlast (7.6 µg/L/hour) was well within an 80%–125% interval of intravenous median AUC0-Tlast (8.7 µg/L/hour, 80%–125% interval=7.0–10.9 µg/L/hour). Cmax and tmax were expectedly significantly different (p=0.014 and p<0.001, respectively).
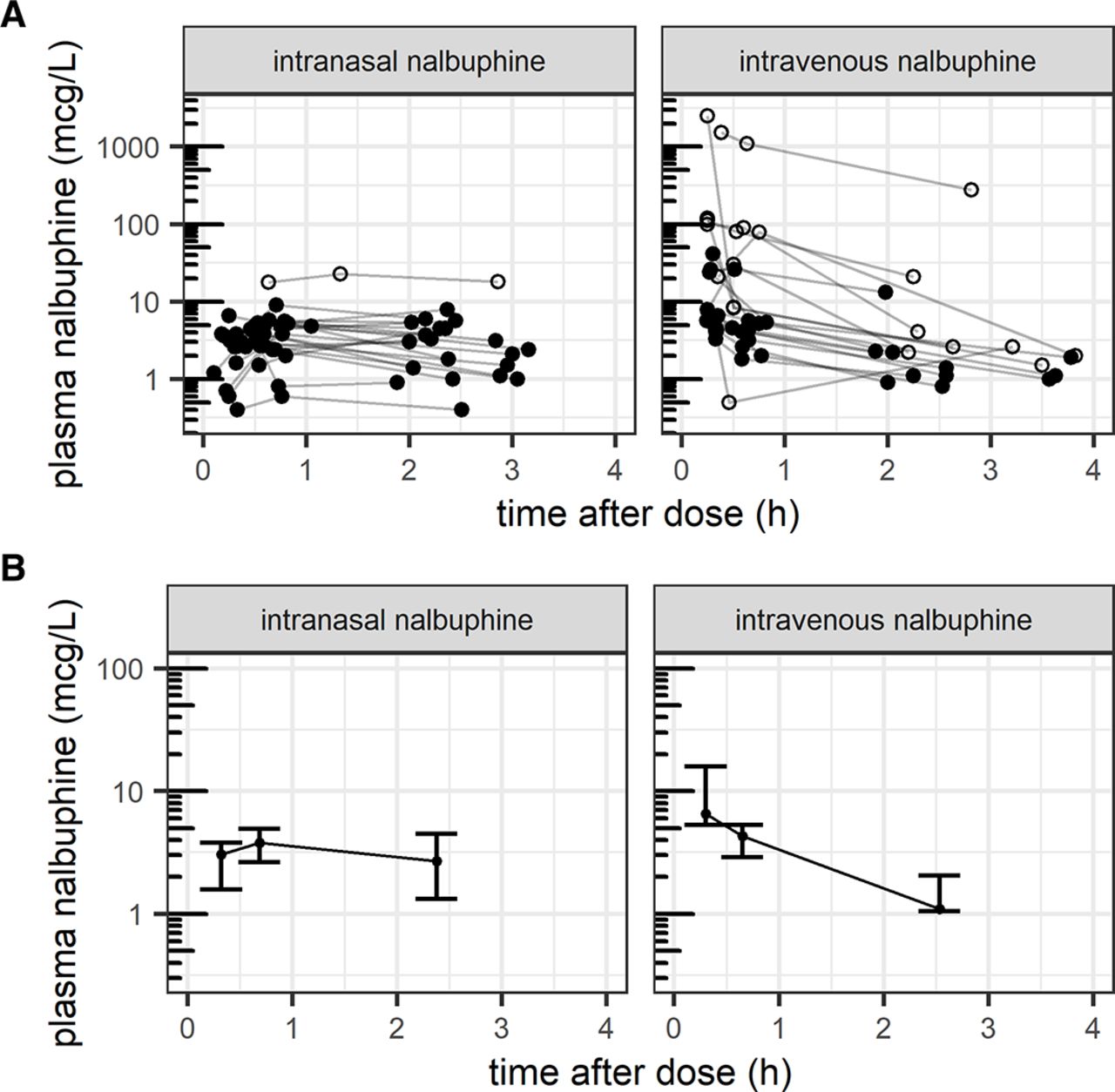
Illustration of (A) measured pharmacokinetic profiles with outliers (open circles) from n=40 patients with all three concentration measurements and (B) summary pharmacokinetic profile showing median and IQR from n=31 patients included in non-compartmental pharmacokinetic analysis.
NCA: pharmacokinetic parameters of nalbuphine after 0.05 mg/kg intravenously or 0.1 mg/kg intranasally
Tolerability of intranasal application and pain control
52 patients received nalbuphine, 35 of them (14 intravenous/21 intranasal) were eligible for tolerability assessment due to available recorded NIPS during drug administration. Mild to no pain was recorded in 71% during intravenous and 67% during intranasal nalbuphine administration.
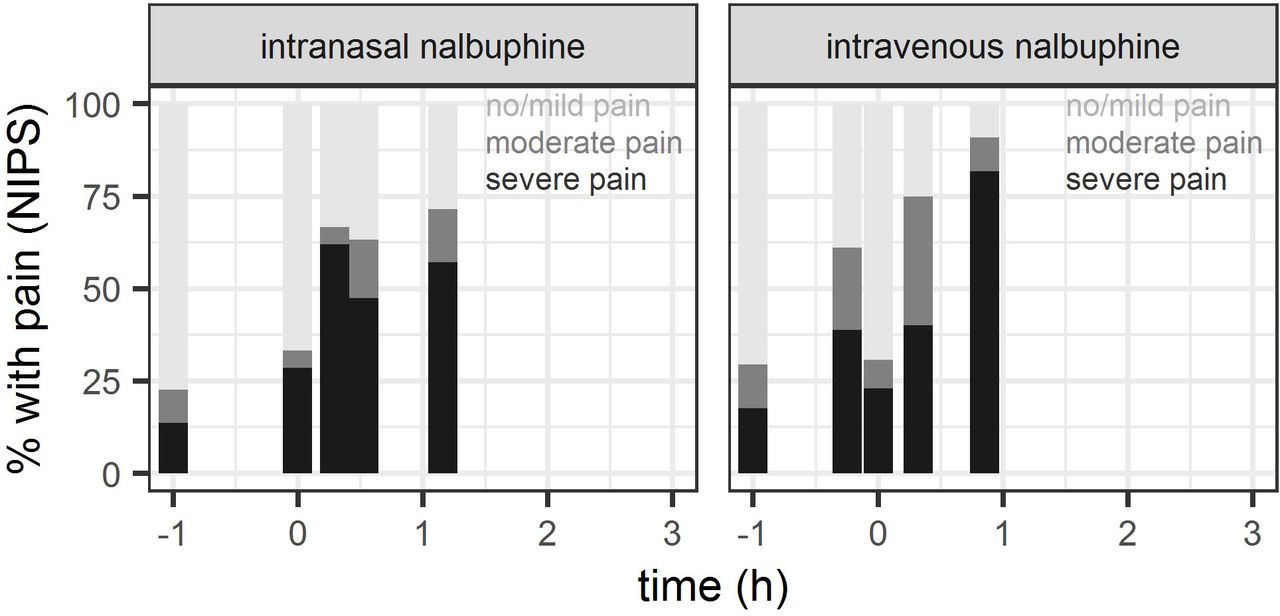
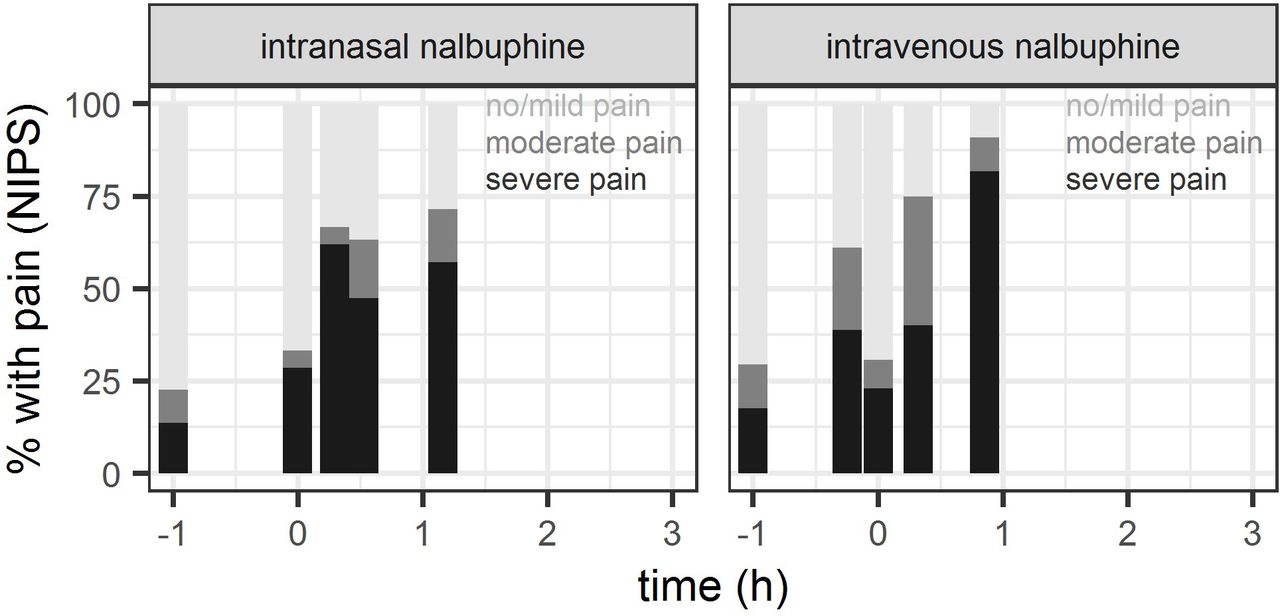
Out of 52 patients having received nalbuphine, recorded NIPS were available for 40 patients (19 intravenous, 21 intranasal) during placement of peripheral intravenous access, 42 patients (22 intravenous, 20 intranasal) during urinary catheterisation and 25 patients (11 intravenous, 14 intranasal) during lumbar puncture. NIPS measurements are summarised in figure 3 (detailed numbers: see online supplemental table). Severe pain was recorded in the intravenous (intranasal) study group during insertion of peripheral intravenous access in 42% (62%), during urinary catheterisation in 45% (50%) and during lumbar puncture in 82% (57%) of study subjects, respectively.
Supplemental material
{kind=link}
{kind=link}
{kind=link}
Proportion of patients with NIPS classified as no/mild pain (NIPS <3), moderate (NIPS=3–4) or severe pain (NIPS >4), evaluated as indicator of pain control during interventions (establishment of venous (intravenous) access shortly before (intravenous group) and shortly after nalbuphine administration (intranasal group), respectively, urinary catheterisation and lumbar puncture) and as indicator or tolerance of nalbuphine administration. Baseline: NIPS assessment in the time between informed consent and first intervention (intranasal nalbuphine administration or venous access) plotted at time=-1h.
Safety (adverse events and vital signs)
Oxygen saturation, heart rate and blood pressure are summarised graphically measured in all 52 patients and as mean change from baseline with 95% CI in online supplemental figure. No measurement required medical intervention.
No serious adverse event was noted. Five mild adverse events, which were assessed related to nalbuphine, were reported: two patients vomited, one patient became pale and two patients were sleepy and dizzy. None of these patients had implausible high serum nalbuphine concentrations. The two patients with documented accidental overdoses had mild to moderate adverse events, which were classified as likely related to nalbuphine by the treating physician. The patient in the intranasal study group suffered bradycardia (6 hours after nalbuphine administration) and self-limiting apnoea. The other patient (intravenous group) slept for an unusually long time.
Discussion
This is the first study reporting clinical PK of intranasal nalbuphine in infants 1–3 months of age. We observed similar median AUC0-Tlast during the first 2.5 hours under studied doses, suggesting an intranasal bioavailability close to 50%. As expected, however, measured median Cmax was lower following intranasal compared with intravenous administration, with tmax having occurred after the 15 min sample only (median measured tmax close to 40 min). These different kinetic profiles may need to be considered for optimal timing of interventions.
Intranasal administration was overall well tolerated by the majority (67%) of patients recorded with mild to no pain only (NIPS <3), despite the low pH of 3.0–4.2 of the nalbuphine solution (the optimum pH for intranasal application is 4.5–6.5).21–24 27 Furthermore, we were able to show that nalbuphine exhibits a good safety profile. No serious adverse event was noted despite two 10-fold overdoses, and generally only few mild to moderate adverse events occurred, which were still deemed related to nalbuphine. The delayed bradycardia observed in one of the patients (noted 6 hours after intranasal nalbuphine overdose) might be related to possible partial intestinal absorption, which is expected to be delayed during opioid exposure due to inhibition of gastric emptying.28
Given the relatively high proportion of patients with severe pain during urinary catheterisation and lumbar puncture (range across groups: 45%–82%), we hypothesise that dosages may need to be increased in practice to usual doses corresponding to 0.1–0.2 mg/kg intravenously,1 compared with our studied doses of 0.05 mg/kg intravenously and 0.1 mg/kg intranasally. A target concentration under continuous infusion of 12 µg/L in children >1 years has been proposed by Bressolle et al 3 for postoperative pain treatment, and was achieved neither by our median measured Cmax of 4.5 µg/L after intranasal administration nor by our median concentration of 6.5 µg/L 15 min after intravenous administration. A complementary population PK/pharmacodynamic analysis of our study data predicts median Cmax of 17.9 µg/L (IQR: 7.5–32.8) after the intravenous dose of 0.05 mg/kg (at t=0 hour), and supports usefulness of intravenous nalbuphine doses of 0.1–0.2 mg/kg to exceed the proposed target concentration of 12 µg/L for at least 30 min following drug administration.29 Consistent with these findings, model-based simulations suggest that the proportion of patients with severe procedure-related pain at 30 min may be reduced from approximately 40% (under 0.05 mg/kg intravenously) to ≤20% (under 0.1–0.2 mg/kg intravenously).29 A comparable achievement of target concentrations and pain control after intranasal use would be expected only at an intranasal dose of 0.4 mg/kg according to model simulations.29
Our initial study doses of 0.1 mg/kg intravenous and 0.2 mg/kg intranasal nalbuphine were not approved by the Swiss health authority due to safety concerns in infants <3 months, although the licensed intravenous dose was 0.1–0.2 mg/kg, without a specified age limit in Switzerland at that time30 (note: dose is limited to children over 1.5 years old now1). The study was performed at our high frequency emergency department (45 000 patients/year) and therefore safety measures differ from intensive care unit or anaesthesia department where continuous monitoring and observation of the patient can be provided. Nalbuphine was newly introduced for procedural pain management in this setting. A ‘low-dose’ nalbuphine was therefore expected to be better than current standard of care (glucose cotton swab, no pain medication) and as such, a careful dosing approach was ethically justifiable.
Limitations
In 40 patients of 52 (19 intravenous/21 intranasal), all three planned nalbuphine concentration measurements were obtained. In the remaining 12 patients, we did not achieve to obtain all needed serum samples due to either loss of venous access or no return of blood from intravenous line. In the protocol, we decided not to insert another intravenous access or perform a puncture for blood sampling just for study purposes. We excluded a total of eight patients in the intravenous group from PK analysis due to implausible serum concentrations partly because rising concentrations were observed, which may be explained by accidental sample switch, and partly due to extremely high concentrations. We assumed that the latter was mainly related to the blood withdrawal, as blood samples were taken from the same intravenous access that was used for nalbuphine administration. If the nurse forgot to flush the intravenous access after intravenous nalbuphine administration, part of the nalbuphine dose may have entered the blood sample for serum concentration measurement, thus achieving an implausible high value. Another point which may have introduced some inaccuracy in dosing was the small drug volume (0.02–0.1 mL) administered. Unfortunately, we had to exclude two patients (one intravenous/one intranasal), due to an accidental (10-fold) overdose. The NIPS measured in this study is an indicator for pain or distress in infants aged less than 1 year, and uses body language to help us to understand if an infant is in pain. At this non-verbal age, infants cannot tell us if they are in pain, so we can use this scale only as a surrogate. It is possible that the NIPS was partly high simply because the infant was hungry, did not like its positioning or felt discomfort, for example, associated with intranasal drug administration. Alternative scores have been advocated for pain assessment in infants (eg, Neonatal Facial Coding System31). In this study, however, the NIPS was used because it is the standard in our emergency department. As our complementary analysis found associations between individual nalbuphine exposure and pain response in terms of NIPS,29 we may conclude that this score was still a suitable measure for pain assessment in our study despite its limitations. NIPS was missing for 15%–58% of the interventions, partly also because the planned intervention was not performed. The lumbar puncture, in particular, was not carried out in all cases (for example, for patients qualifying for partial sepsis work-up). Due to lack of randomisation (use of open alternate allocation procedure), some selection bias cannot fully be excluded. Finally, we did not obtain concentration measurements after 3 hours, limiting assessment of AUC0-infinity after intranasal administration.
Conclusion
This is the first study reporting clinical PK after intranasal and intravenous administration of nalbuphine in infants 1–3 months of age. We observed similar exposure coverage following single administration of 0.1 mg/kg intranasal and 0.05 mg/kg intravenous nalbuphine, suggesting an intranasal bioavailability in the order of 50%. Intranasal administration was safe and well tolerated in 67% of patients. As such, intranasal dosing could be a safe, non-invasive alternative approach to parenteral administration of nalbuphine in clinical practice, especially if establishing a venous access is not feasible or too time-consuming. Intranasal dosing has the potential to reduce pain for paediatric patients and stress for parents and medical staff. Additional studies are warranted to investigate optimal dosing and timing of interventions as Cmax is delayed by half an hour after intranasal administration.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by the local Ethics Committee and the Swiss Agency for Therapeutic Products (Swissmedic) (ClinicalTrials.gov Identifier: NCT03059511). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors would like to thank the whole team of the Children’s Hospital Zurich emergency department for their great commitment. We also thank Martin Volleberg, Institute of Clinical Chemistry, University Children’s Hospital Zurich; Dr Daniel Müller, Institute of Clinical Chemistry, University Hospital Zurich; and Dr Andrew Attkinson, Pediatric Pharmacology and Pharmacometrics Research Center, University Children's Hospital Basel. We also would like to thank the patients and their parents for their participation in this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @PVonbach
MP and VG contributed equally.
Contributors MPfiffner—conceptualisation/design, methodology, data curation, investigation, formal analysis, writing (drafting the initial manuscript), final approval of the version to be published. VG—data curation, formal analysis, supervision/oversight, writing (review or editing of the manuscript), final approval of the version to be published. PV—conceptualisation/design, methodology, funding acquisition, supervision/oversight, resources, writing (review or editing of the manuscript), final approval of the version to be published. MPfister—conceptualisation/design, methodology, funding acquisition, supervision/oversight, resources, writing (review or editing of the manuscript), final approval of the version to be published. EB-O—conceptualisation/design, methodology, data curation, investigation, supervision/oversight, resources, writing (review or editing of the manuscript), final approval of the version to be published.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.