Article Text

Abstract
Objective Clinical trial sponsors spend considerable resources preparing informed consent (IC) and assent documentation for multinational paediatric clinical trial applications in Europe due to the limited and dispersed patient populations, the variation of national legal and ethical requirements, and the lack of detailed guidance. The aim of this study was to design new easy-to-use guide publicly available on European Medicines Agency’s, Enpr-EMA website for all stakeholders.
Methods Current EU legal, ethical and regulatory guidance for paediatric clinical trials were collated, analysed and divided into 30 subject elements in two tables. The European Network of Young Person’s Advisory Group reviewed the data and provided specific comments. A three-level recommendation using ‘traffic light’ symbols was designed for four age groups of children, according to relevance and the requirements.
Results A single guide document includes two tables: (1) general information and (2) trial-specific information. In the age group of 6–9 years old, 92% of the trial-specific subject elements can be or should be included in the IC discussion. Even in the youngest possible age group (2–5 years old children), the number of elements considered was, on average, 52%.
Conclusion The EU Clinical Trial Regulation (2014) does not contain specific requirements exclusively for paediatric clinical trials. This work is the first to extensively collate all the current legal, regulatory and ethical documentation on the IC process, together with input from adolescents. This guide may increase the ethical standards in paediatric clinical trials.
- ethics
- therapeutics
Data availability statement
Data are available in a public, open access repository. All data are publicly available via Enpr-EMA website: https://www.ema.europa.eu/en/documents/other/assent/informed-consent-guidance-paediatric-clinical-trials-medicinal-products-europe_en.pdf.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
What is already known on this topic?
Paediatric clinical trial sponsors spend considerable resources preparing informed consent (IC) and assent documents for multinational paediatric Clinical Trial Applications in Europe.
There are no detailed and easy-to-use public guide for designing of IC and assent forms for paediatric multicentre clinical trials.
Many national legal and ethical guidance still lack practical common advice for design of IC documents for children and ensuring children’s involvement.
What this study adds?
This guide provides practical instructions for legal and ethical requirements for IC and assent for children of all age groups.
This guide includes input from children, providing ideas on how to enhance the involvement of children in the IC process.
This guide supports sponsors’ Clinical Trial Application submissions to Competent Authorities and Ethics Committees, enhancing the high-quality ethical standards of paediatric clinical trials.
Introduction
Children represent 20% of the European population (~90 million citizens).1 In 2020, on average, 11% of all clinical trials registered into the European Clinical Trials database (EudraCT) were paediatric clinical trials with investigational medicinal product.2 Due to the EU Paediatric Regulation,3 all new clinical trials with medicinal products must consider if the inclusion of children (0–18 years of age) from all or some age ranges is relevant, which means the number of paediatric clinical trials will increase. The number of participants per country can be very small.
These multicentre and multinational trials create demanding research environment for academic groups and pharmaceutical industry in Europe, as they spend considerable time and resources preparing documentation for clinical trial application. The submission packages for the competent authorities and ethics committees including informed consent (IC) and assent documents are challenging due to non-harmonised legislation and wide variation between national regulatory requirements.4 5
In Europe, the legal age for giving independent IC for participation in a clinical trial varies between 14 and 18 years. In paediatric trials, the consent process includes the child’s own consent, assent or agreement. According to many national laws, a child’s own assent is usually not sufficient alone to allow his/her participation, unless supplemented by the consent of the child’s legally designated representative(s).4 6
Legal, ethical and regulatory framework for paediatric clinical trials in Europe
After the EU Paediatric Regulation (EC No 1901/2006),3 in 2014 the EU Clinical Trials Regulation followed7 the current EU Clinical Trial Directive.8 The clinical trials regulation will facilitate clinical trials in the EU after it will come into application on 31 January 2022. Under this regulation, ethical review will remain under each EU Member State according to their national laws. In May 2018, the new EU General Data Protection Regulation9 came into force, impacting clinical trials and consent process, requiring an explanation of the legal basis for the collection and processing personal data on trials. Some countries require even explicit consent for data processing.
The ethical foundation for clinical research is the updated World Medical Association’s Declaration of Helsinki (WMA DoH),10 incorporated into the International Conference on Harmonisation (ICH) Good Clinical Practice (GCP) guideline.11 Both principles of the WMA DoH and GCP are evident in the EU Clinical Trial Regulation. In 2017, the EU Commission’s revised version of the EU ethics guideline for paediatric clinical trials was published.6
Children’s right to be involved in clinical trial design and IC process
According to the current legal texts and ethical recommendations, children should be able to take part in the consent process. A child’s participation is derived from the general requirements for the right to express their own opinions from the Fundamental rights of the Convention on the Rights of the Child.12 13
The increased patient and parent involvement have evolved after the European Young Person’s Advisory Groups (eYPAGnet) was established in 2017.14 The eYPAGnet is a consortium of Young People’s Advisory Groups (YPAGs) across Europe, supporting the development of new national YPAGs. The YPAG provide a platform for children to have a voice, share their opinions and apply their experience to a variety of issues in clinical trials, such as ICs and assents. These groups include young people aged between 8 and 19 years (even up to 21 years) who are patients and/or healthy children having an interest in science, healthcare and children’s rights.15
Comprehensive trial information must always be presented to the child’s legal representative(s) both orally and in writing.6 Trial information should be also provided to the child participant, but it should be adapted to the child’s language skills and understanding, the child’s developmental stage, intellectual capacity, medical condition, previous life/disease experience and other circumstances. Chronological age only partly correlates with maturity. The maturity is evaluated by the investigator. Where appropriate, translation should be arranged. It is very important to identify a child’s potential dissent or disagreement to participate, which should always be respected.6
Enpr-EMA facilitates the main objective of the paediatric regulation
In 2013, the European Network of Paediatric Research at the European Medicines Agency (Enpr-EMA) initiated various multistakeholder working groups to find solutions to emerging medical needs related to paediatric clinical trials.16–18 The ethics working group has focused on ethical issues in paediatric clinical trials and published first ‘Tool Kit’ of European informed consent and assent requirements in December 201519 and related article later in May 2016.4
The aim of the new guide for paediatric ICs and assents
Some EU-funded projects and learnt societies have developed new practical tools, guidance, design methods and training programmes for supporting paediatric clinical trials.20–25 The aim of this new guide was to provide more practical easy-to-use tool for designing the content of IC and assent documents to enhance the high-quality ethical standards of paediatric clinical trials. This guide can be adapted to all types of paediatric clinical trials on a case-by-case basis, and it can also be used to promote the involvement of children in the consent process. This guide was initially published on Enpr-EMA website on January 2021.26
Methods
The working group collated all current EU legislative, ethical and regulatory legal texts specific to paediatric clinical trials via literature search and used these as the primary data source.
A total of 30 identified main subject elements required for the consent process in legal, ethical or regulatory texts were divided into two tables based on the nature of the requirement. The first table is applicable to all trials (5 general elements) and the second table includes trial-specific topics (25 elements), which can vary between trial design (table 1). All subject elements were considered for four paediatric age groups (0–2; 2–5; 6–9; 10–18) and legal representative(s) as defined in the EU ethics guideline.6
Subject elements of IC and assent guide tables 3 and 4
A three-level recommendation was established as ‘traffic light’ symbols. These were used to separate subject elements to be considered, not considered or categorised as optional according to the age group per each subject element (table 2).
Three-level recommendation symbols for all age groups
The three-level recommendation was reviewed against each subject element (5+25 elements) and across all age groups. All elements were sorted with the symbols to separate each subject element according to suitability for each age group and this resulted in recommendations per age group.
All subject elements were listed separately as these are unrelated to each other. The data were then arranged into five vertical main sections (columns) (I–V): (I) age group in years, (II) legal representative(s), (III) elements to consider/information which must be included in the assent/consent document, (IV) questions to be addressed, and (V) notes and example methods/texts to be used. The first heading (I) includes four age group columns. One additional sixth section (column VI) was added to table 4 for the element numbering (tables 3 and 4). Both tables include text partly written in bold, emphasising the most important aspects, or requirement, need to be considered.
General information for informed consent and assent (agreements)
Trial specific information for informed consent and assent (agreements)
After structural design, the eYPAGnet members of three national YPAGs (the UK, Scotland and Spain) were requested to review all 30 subject elements in order to identify important information, any issues or missing information and the preferred format of given information for a practical consent discussion. All comments were collected by monographic sessions led by the group facilitators. All feedback was collated into the report. The report data were incorporated into the guide’s section IV, as questions, example methods or texts to be used when designing consent/assent documents, or when recruiting paediatric participants to trials.
The last review for the complete guide was done by the Office of the Paediatric Medicines (Scientific Evidence Generation Department) at the European Medicines Agency. The guide document was finalised according to these comments (figure 1).

The flowchart of the development process of the informed consent and assent guide.
Results
Subject elements adaptable and suitable for various age groups is very high
After all the subject elements were collated, it clearly illustrated the complexity of IC forms and the consent process in the paediatric population. Many items must be discussed in an adapted manner according to their age and maturity representing all possible therapeutic areas and diseases, from healthy subjects (eg, vaccine trials) to extremely rare disease patient groups.
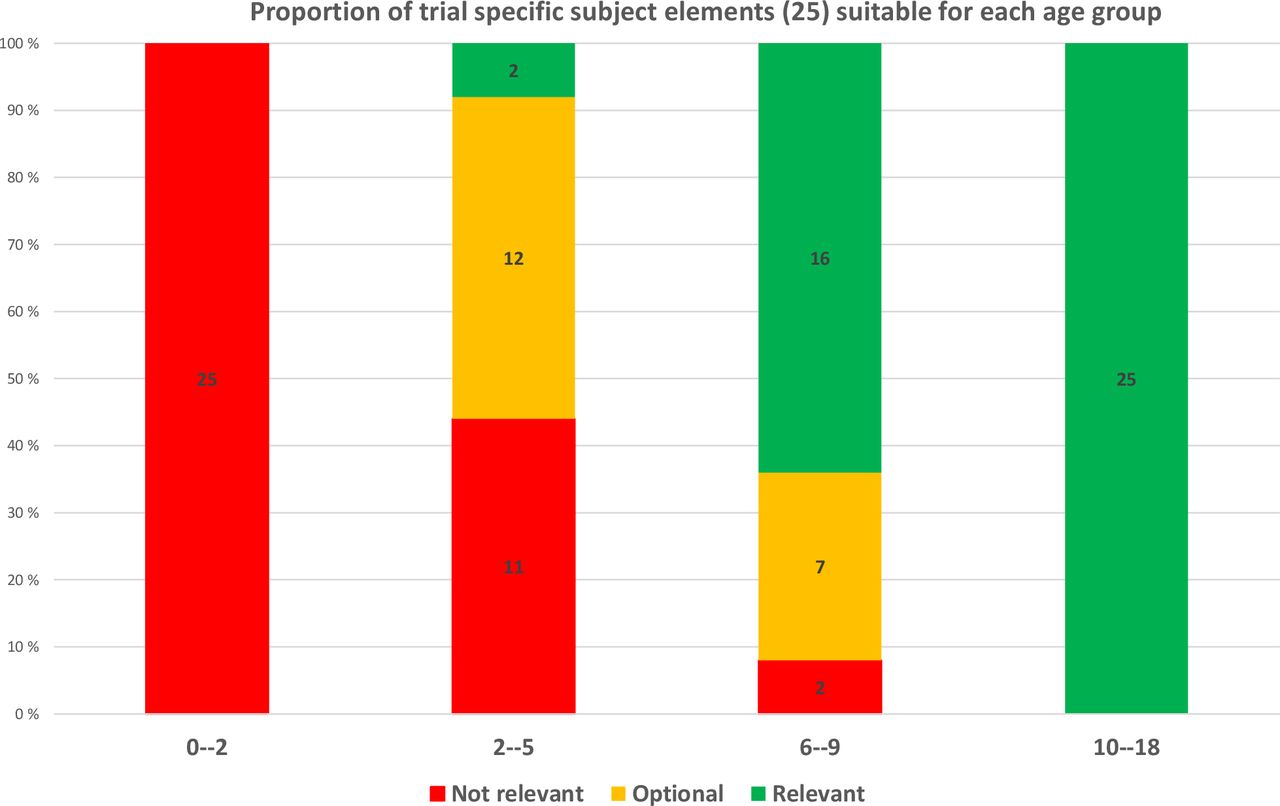
Listing trial-specific subject elements (25 elements of table 4) relevant to all paediatric clinical trials resulted in all elements (100%) being relevant and to be considered when designing and conducting clinical trials in the oldest age group. Further, when looking at children in the age group 6–9 years, 23 out of 25 (92%) elements can be optionally included or should be included in adapted ways in the consent process. Even in the youngest possible age group, the number of optional or recommended elements is still high; 12–14 out of 25 (on average 52%). The inclusion of small children (under 2 years) is discussed only with the legal representative(s) (figure 2).
{kind=link}
{kind=link}
Proportion of information of 25 trial-specific subject elements relevant to each four age groups (red=not relevant, orange=optional, green=relevant).
The specific questions addressed to adolescents in the consent discussion
The comments received from the eYPAGnet members resulted in a high number of recommended questions (81) to adolescents per each subject element. The total number of questions in both tables (3 and 4) is 86, and the number of questions varies between one and six per subject element. The first five questions to the table 3 were generated by the working group.
Discussion
The practical challenges in the implementation of paediatric multicentre clinical trial
Paediatric clinical trial design and recruitment need specific expertise and experienced, qualified and trained personnel. Similar experience is required from the people reviewing and assessing the trial protocols, patient information, consent and the assent or agreement forms.6
Pharmaceutical companies and Contract Research Organisations have their own document templates, resulting in hundreds of different consent and assent documents submitted to European ethics committees, which repeatedly receive incomplete or inappropriate documentation for paediatric clinical trials. The most problematic area is patient information and consent forms, as these are often too complex for children to understand, and too extensive to read and comprehend. The language is often targeted for adults. The child’s participation cannot rely on the adult IC, which applies to decisions made by those with the legal and intellectual capacity to make such choices. Children usually lack such capacity, and they need adapted information.
Repeated amendments and committee’s re-evaluations prolong ethics approvals and cause delays to the start of clinical trials, causing unnecessary loss of time and money. To avoid such pitfalls, consent and assent documents need to be designed according to high ethical standards.6 27–31
The age groups of children can be used for different purposes depending on scope
The International Conference of Harmonisation (ICH) E11 Guideline32 lists five age groups (preterm newborn infants, term newborn infants; 0– 27+ days, infants and toddlers; 28 days to 23 months, children; 2–11 years and adolescents; 12 to 16–18 years dependent on the region). This ICH guideline is scoped for trial design, covering, for example, pharmacological assessment and outcome measures based on physiological development and pathophysiology. Four age groups used in this consent and assent guide document (0–2; 2–5; 6–9; 10–18 years) are those described in the EU ethics guideline6 and differ slightly from the ICH guideline, as the purpose and scope is to provide general instructions for planning the consent process and the appropriate involvement of children.
New European regulations affect the IC process
For the first time, the EU Clinical Trial Regulation makes the involvement of children in the consent process mandatory, according to age and maturity.7 Under the EU General Data Protection Regulation (GDPR),9 individuals normally gain the right to be asked for consent (or to withhold consent) for the collection and processing of their personal data between the ages of 13 and 16 years. In a clinical trial setting, where the age of consent to participate is generally older (14–18 years), this may create discrepancy. Recital 161 of the GDPR stipulates that the age of consent to personal data processing will be the same as the age to consent for clinical trial participation.9
However, it is important to distinguish between the requirement for consent for clinical trial participation and the requirements for lawful processing of personal data under the GDPR. The standards of quality (reliability of data) and patient safety for medicinal products fall under different legal bases. Therefore, the requirement for IC by the EU Clinical Trial Regulation is a safeguard, not a legal basis for data processing, and must not be confused with consent as a legal ground for processing personal data set out in Article 6(1)(a) of the GDPR.33
The young people provide important expertise to paediatric trials
For this guide, the young person’s groups provided important input. This represents the direct voice of young people and is extremely valuable in the consent discussion. The importance of the opinions and experiences of children and their families is now becoming a crucial part in the development of new medicinal products, as recognised by many stakeholders.34–36
Current existing guidance is not enough to design consents and assents for children
To date, there are only a limited number of public ethical and practical guidance documents regarding consent and assent design specifically for paediatric clinical trials. Some learnt societies have provided articles, books, videos, and recommendations, practical guidance and training programmes for all stakeholders.20–25 However, none of these existing materials provide similarly detailed instructions for the design of the paediatric consent, assent or agreement document structure, and guide for the whole consent process, including the voice of children.
Practical limitations and implementation of this guide
Each Member States of the EU and European Economic Area has its own national legislation and detailed requirements for consent documents,4 and these vary among countries. Therefore, this guide should be used together with the local or national requirements. It is also important to acknowledge that while this guide contains instructional text (ie, ‘should be’), these are not official requirements or legal guidance, but suggestions serving as an example for every age group. All the subject elements need to be adapted case-by-case according to the nature of the clinical trial protocol.
This guide is designed to support the consent process for paediatric clinical trials in Europe across all age groups. It can be used when preparing submission packages to competent authorities and ethics committees. This guide provides practical ideas and examples about how to present relevant information to children in the consent process, and it serves also as an example to encourage to seek comments from the local patient and parent groups when designing consent and assent documents. The content is also applicable to electronic consent. The document will be updated by the Enpr-EMA secretariat and it will be open for public comments for further improvement.
Conclusion
This work is the first to extensively collate and document all the current legal, regulatory and ethical guidelines on the consent and assent process for paediatric clinical trials in Europe, together with input from adolescents. It is a single easy-to-use publicly available guide for all stakeholders and the contents can be adapted to trials in case-by-case.
Although the new EU Clinical Trial Regulation will harmonise Clinical Trial Application practices in Europe, it does not contain specific guidance for the paediatric clinical trial consent process. As there are still many national legal differences in the requirements for consent and assent documents, this guide may increase the level of ethical standards and facilitate the harmonisation of paediatric consent and assent documents in Europe.
Data availability statement
Data are available in a public, open access repository. All data are publicly available via Enpr-EMA website: https://www.ema.europa.eu/en/documents/other/assent/informed-consent-guidance-paediatric-clinical-trials-medicinal-products-europe_en.pdf.
Ethics statements
Patient consent for publication
Acknowledgments
Special thanks go out to eYPAGnet group leaders; Jennifer Preston, GenerationR (England), Pamela Dicks, ScotCRN (Scotland), Begonya Nafria Escalera, Kids Barcelona (Spain) and Segolene Gaillard, Kids France (France) and their great effort when collecting comments from all national adolescent groups giving valuable add to this guideline.
Footnotes
Contributors Literature search and guide document table design was done by PL, MK, VG, HG, MD-K, HD, DN, GBB, JC, JD and DH. Data figures and tables to the article were done by PL. Data interpretation, writing and approval of final version were done by all authors. Guarantor: Pirkko Lepola (PL)
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests PL is the Chair of the Coordinating Group of the European Network of Paediatric Research at the European Medicines Agency (Enpr-EMA), and all authors have been members of the ad hoc Enpr-EMA Ethics Working Group during this work.
Provenance and peer review Not commissioned; externally peer reviewed.