Article Text

Abstract
Objective To retrospectively investigate the clinical spectrum, genetic profiles and outcomes of survivors of paediatric sudden cardiac arrest (SCA).
Design and patients All 66 patients (aged 1–20 years), with unexpected SCA or syncope related to ventricular tachycardia (VT)/fibrillation and who survived to discharge from a tertiary centre, were enrolled from 1995 to 2018. Of these, 30 with underlying diseases prior to the events were excluded. Whole-exome sequencing targeting 384 channelopathy and cardiomyopathy-related genes (composite panel) was conducted to identify the possible genetic variants/mutations.
Results A total of 36 patients were enrolled. Male adolescents predominated (66.7%), and the median age at onset was 13.3 years. Events occurred most often during exercise and daily activities. The yield rate of the genetic test was 84.6% (22/26); 14 had pathogenic variants; and 8 had likely pathogenic variants. The most common diagnoses were long QT in nine (25%), catecholaminergic polymorphic VT in six patients (16.7%), but other long QT and cardiomyopathy genes were also detected in eight patients (30.7%). The 10-year transplantation-free survival rate was 87.8% and was better for those who received genetic tests initially at the disease onset. An implantable cardioverter–defibrillator was implanted in 55.6% of the patients, with an appropriate shock rate of 61.1%. The defibrillator shock rate was lower for those who received composite panel initially.
Conclusion Survivors of SCA in the paediatric population had favourable long-term outcomes aided by genetic test. A broad composite genetic panel brings extra diagnostic value in the investigation of ventricular fibrillation/sudden cardiac death.
- cardiology
- genetics
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Statistics from Altmetric.com
What is already known on this topic?
Short-term outcomes improved with the introduction of bystander cardiopulmonary resuscitation and the automated external defibrillator.
A genetic test can help identify underlying channelopathy or cardiomyopathy in survivors of paediatric sudden cardiac arrest (SCA).
The yield rate of genetic test for survivors of SCA was low even with conventional genetic panel studies.
What this study adds?
The long-term outcome of survivors of paediatric SCA is good after an aggressive intervention.
RYR2 and common long QT (LQT) syndrome genes are the most common causes, but other minor LQT syndrome genes and cardiomyopathies genes are also crucial.
The novel state-of-the-art composite panel including channelopathy and cardiomyopathy genes increased the yield rate and may modify the treatment and improve outcome.
Introduction
Sudden unexpected death (SUD) is an important but uncommon cause of death in the young population. In this population, the incidence of SUD is 2.7 per 100 000 person-years in Taiwan, which is similar to that in Western countries.1–3 This drastic presentation and the impact on the family and community make it an important public health issue. Among the causes of SUD, >30%–50% of cases are attributed to cardiac causes.3 The outcomes of children with sudden cardiac arrest (SCA) are poor, with a survival rate of only 10.4%.4 The survival rate of SCA has increased to 40% due to bystander cardiopulmonary resuscitation (CPR), public automated external defibrillator (AED) use and effective emergency medical service responses.1 5 6
The common causes of SCA in the young population include structural heart disease, hereditary arrhythmia syndrome and coronary artery anomalies.1 3 7 ECG and echocardiography are effective screening tools, and imaging studies, such as cardiac MRI and electrophysiological studies, are effective diagnostic tools for these patients.3 8 Nevertheless, the aetiology of SCA remains unknown in many of these patients.9 Autopsies have revealed negative results in up to 50% of patients with SUD.1 3 Molecular genetic studies have provided clues of possible aetiologies in these patients.10
Several studies have focused on genetic testing for the decedents of SUD.1 3 9 11 However, relevant studies involving genetic testing in survivors of paediatric SCA are rare.6 In this study, we studied the impact of genetic testing conducted using a state-of-the-art composite genetic panel on the survivors of paediatric SCA.
Patients and methods
Patient selection
This study identified all patients aged 1–20 years who were sent to or referred to National Taiwan University Hospital and had a diagnosis of (1) unexpected SCA out of hospital or within 24 hours after admission, or (2) witnessed syncope whose first ECG either in the ambulance or in the emergency department showed ventricular tachycardia (VT) between January 1995 and December 2018. Only those who survived to discharge were enrolled. SCA was defined as malfunction or cessation of the electrical and mechanical activities of the heart, resulting in near instantaneous loss of consciousness and collapse.1 Patients with obvious causes of arrest, such as trauma or accident, were excluded. After initial screening through chart review, 78 potential patients were selected. Of these, 12 were excluded due to age of <1 year, and 30 other patients were excluded because of structural heart disease or severe underlying diseases (23 with known congenital heart diseases or cardiomyopathies before events, 5 with severe neurological diseases and 2 for other reasons).
Demographic data and all medical records, including previous genetic study results (mostly using the candidate gene approach) were collected, and the patients were invited for a further genetic study. A good neurological outcome was defined as a normal neurological examination and no need for special education during follow-up.
Mutational screening and analysis
Genomic DNA was isolated from the venous blood of the patients using standard procedures. A candidate gene approach was performed as an adjunct to clinical diagnosis between 1995 and 2010. In 2011, we designed a state-of-the-art composite genetic panel which included hereditary arrhythmia and cardiomyopathy genes (online supplemental table 1), and we invited these survivors of SCD back for this genetic panel study.12 We included genes recommended by Heart Rhythm Society/European Heart Rhythm Association/Asia-Pacific Heart Rhythm Society expert consensus13 and genes associated with hereditary arrhythmia and cardiomyopathy, as reported in the literature, ClinVar and the Human Gene Mutation Database (professional version). In total, 384 channelopathy and cardiomyopathy-related genes were screened using next-generation sequencing and MiSeq (Illumina, San Diego, California, USA). The procedures included capture-based exomic sequencing, variant calling and annotation and were performed as we described previously.12 Moreover, we performed in silico analyses using six algorithms: REVEL,14 CADD (http://cadd.gs.washington.edu/home),15 SIFT (http://sift.jcvi.org/www/SIFT_enst_submit.html), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), FATHMM and MutationTaster. A CADD PHRED score of >15 or a REVEL score of >0.50 was used to define deleterious mutations; both are ensemble methods integrating several prediction models. Genomic Evolutionary Rate Profiling (GERP++) was used to estimate evolutionary constraint, in which an RS score of >2 indicated a constrained site. We used the Taiwan Biobank for the in-house control.16 For candidate variants with an allele frequency of <0.001 according to the Taiwan Biobank, we followed the definition of the American College of Medical Genetics and Genomics and considered it a pathogenic variant if it was either a null variant (nonsense or frameshift) or classified as pathogenic in ClinVar or Human Gene Mutation Database, and consider it as a likely pathogenic variant if it did not meet the criteria above but was predicted as deleterious by six in silico analyses, along with a high RS score in the GERP++ as in our previous work.17 Finally, Sanger sequencing was used to confirm the nucleotide changes in the identified variants.
Supplemental material
Statistical analysis
Data are expressed as mean±SD. The non-parametric Mann-Whitney U test was used for the numerical data analysis, and χ2 or Fisher’s exact test was used for the categorical data analysis. The Kaplan-Meier survival curve was used for the survival analysis. A p value of <0.05 was considered significant.
Results
A total of 36 patients were enrolled: 30 patients with SCA and six in whom syncope with VT was documented at the first recovery ECG. The demographic data are presented in table 1. Male patients were predominant (66.7%). The median age of onset was 13.3 years. Most of the events occurred during exercise or daily activities. All cardiac events were directly witnessed and mostly by classmates or teachers. Most events occurred at school (30.6%) or in a public place (33.3%). Bystander CPR was performed in 30 (83.3%) of the patients, and onsite AED or defibrillator shock therapy was provided to 19 (52.8%) patients. Positive family history of SCA or arrhythmia-related syncope within first-degree relatives was found in 9 of 36 (25%) patients (online supplemental table 2).
Demographic data (N=36)
Genetic study
The results of genetics test and outcomes are summarised in figure 1. Using the initial candidate gene approach, we found that only four (28.5%) patients showed positive results for genetic testing. Eight of the other 10 patients had positive genetic results after the composite genetic panel study provided by this study (figure 1 and online supplemental table 3). Of the 22 (84.6%) patients with a positive genetic diagnosis, according to the aforementioned criteria, 14 had disease-causing pathogenic variants, and 8 had likely pathogenic variants which was deemed to be disease-causing. The yield rate of the genetic study was not different for patients older and younger than 10 years of age (85% vs 100%, p=0.373). In addition, 11 of 26 (42.3%) patients also had a variant of unknown significance and 5 of 26 (19.2%) had likely benign variants on the genetic test. Ten of the 36 patients had not received genetic tests due to either the genetic test being unavailable in the early era or patients/families refused genetic tests. The target gene screening for first-degree family members is shown in online supplemental table 2. For these 26 patients, 9 of the family members refused to receive genetic test, and two had an index family member who died before this event. In the remaining 15 patients, 8 had a positive genetic test in at least one of the family members, and the other 7 patients belonged to de novo mutation.

Illustration of the initial genetic test, retest and outcome of the survivors of SCA.  indicates mortality or heart transplant;
indicates mortality or heart transplant;  indicates appropriate implantable cardioverter–defibrillator shock. SCA, sudden cardiac arrest.
indicates appropriate implantable cardioverter–defibrillator shock. SCA, sudden cardiac arrest.
In the 26 patients who received genetic testing, 16 (61.5%) had positive phenotypes during serial electrophysiological and imaging studies before the genetic test (detailed information in online supplemental table 2). The diagnosis was changed in one patient (patient No 21, from catecholaminergic polymorphic ventricular tachycardia (CPVT) to long QT type 7) after the genetic test. The other 10 patients did not show any positive phenotype during the serial exam, and the diagnosis was only made after the genetic test in eight patients. Two patients of hypertrophic cardiomyopathy (patient No 8 and 14) had aborted SCA as initial presentation. The initial echocardiography showed only mild left ventricular hypertrophy. Early composite panel genetic study revealed MYH7 pathogenic variant, and beta blocker were prescribed. They showed no appropriate implantable cardioverter defibrillator (ICD) shock during follow-up, but their left ventricular mass index doubled at latest follow.
The final diagnosis after the genetic panel study is summarised in table 2. Long QT syndrome was the most common cause of SCA, accounting for 25% (n=9) of the patients. However, only four patients had long QT types 1, 2, and 3. The second most common cause was CPVT, accounting for 16.7% of the patients. Moreover, Brugada syndrome, cardiomyopathy as arrhythmogenic right ventricular cardiomyopathy, hypertrophic cardiomyopathy, and left ventricular non-compaction (LVNC) were not uncommon.
Identified causes in the survivors of sudden cardiac arrest
Outcome
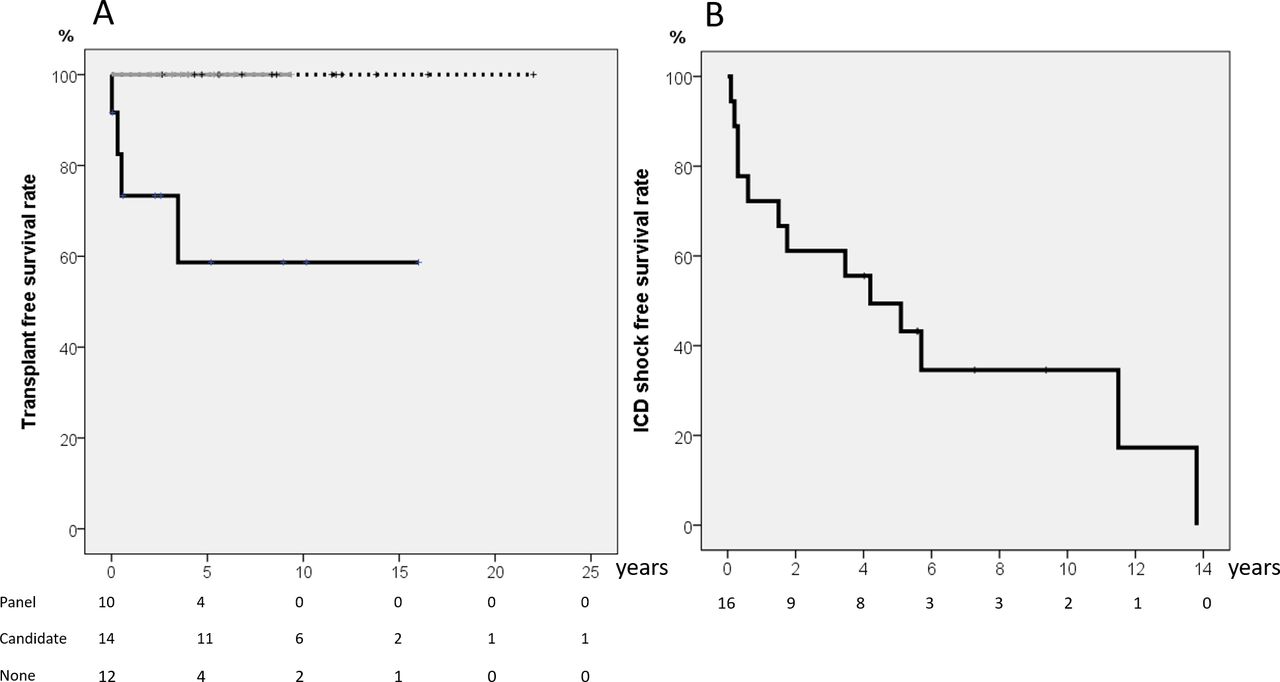
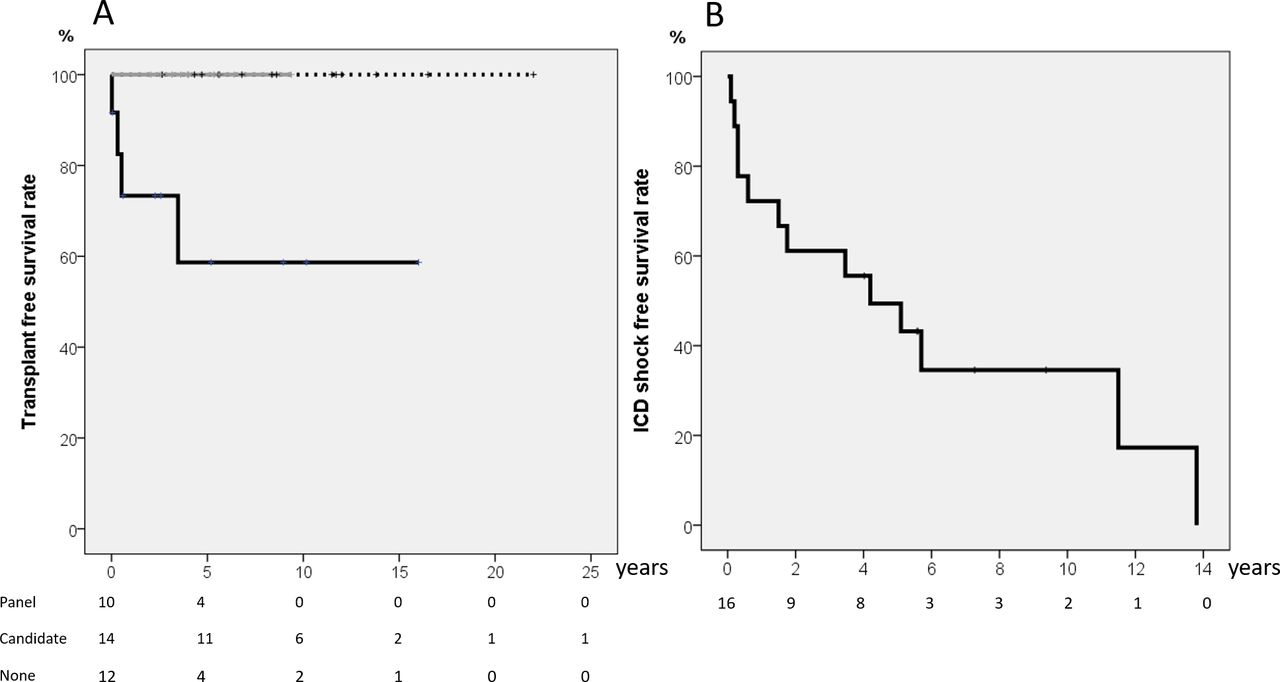
Of the 36 patients who survived to discharge after the SCA events, 2 died during follow-up. One refused ICD implantation and died 7 months after another SCA event. The other patient died of complications from severe neurological sequelae 3 months later. Two patients with LVNC and SCA received an ICD first and then underwent heart transplantation 3 months and 3 years after the event, respectively. After 6.4±5.2 years of follow-up, the heart transplantation free survival rate of the 5 and 10 yearwas 87.8%. All 26 patients who received genetic test initially at the disease onset were alive at the last follow-up. Their heart transplantation free survival rate was better compared with those who did not have any genetic test (figure 2A, p=0.005).
{kind=link}
{kind=link}
(A) Heart transplant free survival curve for all 36 patients categorised according to the initial genetic test. Both of those receiving composite genetic panel and candidate gene approach had higher survival rate (100%) than those without genetic test (p=0.005). Grey line indicates those receiving extended state-of-art genetic panel; dash line indicates those receiving initial candidate gene approach; black line indicates those not receiving genetic test. (B) ICD shock-free survival rate in 20 patients undergoing ICD implantation. ICD, implantable cardioverter–defibrillator.
An ICD was implanted in 18 patients, and 11 (61%) had experienced appropriate shocks (online supplemental table 3). Excluding four patients lost to follow-up and three patients for whom an ICD was not considered mandatory (one with Wolff–Parkinson–White syndrome and two with acquired long QT (LQT) syndrome), there were seven patients who survived without ICD implantation due to personal reason. The ICD shock-free survival curve is shown in figure 2B, showing that 50.6% of the patients would suffer from an appropriate ICD shock 5 years after ICD implantation. The appropriate shock rate was the lowest in those receiving initial composite genetic panel compared with those receiving initial candidate gene approach or no genetic test (20% vs 90% vs 100%, p=0.009). Patient No one had negative genetic result initially under candidate gene approach, and ICD appropriate shock still occurred under beta blocker therapy. The composite genetic panel later showed RyR2 pathogenic variant, and we then added flecainide. She had no more appropriate shock after adjusting medication. Apparent neurological sequelae were found in four patients, including two who died during the follow-up.
Discussion
The present study provides the outcomes and genetic data of survivors of paediatric SCA. The long-term outcomes were favourable with 10 year transplantation-free survival rate up to 87.8% in the current era with aggressive interventions and genetic testing. A state-of-the-art composite genetic panel, which included all currently known hereditary arrhythmia and cardiomyopathy genes, increased the diagnostic yield rate to 84.6% and further modified the treatments.
Genetic study of survivors of SCA
For survivors of SCA, even with thorough investigation, the underlying cause may remain unknown in up to 50% of patients.3 9 Genetic testing has become a critical adjunct to clinical diagnosis. It may not only help to make a definitive diagnosis but also help to guide the medical therapy and life modifications for the patient. The candidate gene approach using Sanger sequencing had been recommended as the standard genetic test in patients with SUD. Commonly examined genes include long QT syndrome and CPVT genes. Anderson et al showed that the candidate gene approach detects 34% of patients with SUD.10 In recent years, next-generation sequencing has become a powerful tool that renders whole-exome sequencing possible. Several reports have shown the effectiveness of next-generation sequencing for the SUD diagnosis in autopsy cases (table 3). The low detection rate in these patients with SUD may be multifactorial. SUD in many infant patients may be related to an unsafe sleeping environment and are not of cardiac origin. Furthermore, neurological deficits as well as hippocampal maldevelopment may be an important cause of SCA in early childhood, particularly in infants.18 Coronary artery anomalies have become the main cause of SUD for older patients, especially those >25 years of age.
 Comparison of whole-exome genetic study results for sudden unexplained death or SCA after 2016
Comparison of whole-exome genetic study results for sudden unexplained death or SCA after 2016
Studies about genetic tests in survivors of SCA are rare (table 3). Giudicessi and Ackerman performed a comprehensive cardiac evaluation. The yield rate of the genetic test was 24.4% in adult SCA patients and up to 75.5% in paediatric population.19 Song et al studied 19 survivors of SCA in an Asian cohort using next-generation sequencing.20 The diagnostic yield rate was 26.3% with another 63.2% of variant of unknown significance.
In the present study cohort, we performed comprehensive electrophysiological and imaging studies as initial investigations similar to the current guideline for all patients.21 22 For the genetic test, we initially performed candidate gene approach based on these study results, which involves screening for possible genes based on clinical suspicion, and found a fair but not high diagnostic yield rate (38%), similar to a previous report.10 However, using next-generation sequencing to screen 384 channelopathy and cardiomyopathy genes with a newly designed state-of-the-art composite genetic panel, we increased the diagnostic yield rate to 84.6%: 64% were pathogenic variants, and 36% were categorised as likely pathogenic. Such a result suggests improved efficacy using such a state-of-the-art composite genetic approach (table 3). The high diagnostic yield rate was explained by the nearly 400 genes that were examined in the present study, which increased the possibility of positive genetic findings. Second, all of our patients who had received genetic testing had VT or VF as their initial rhythm. Third, the age of our cohort ranged between 1 and 20 years. All of these factors increased the possibility of cardiac-origin SCA in these patients.
Genes involved in survivors of SCA
In the present study, the main genetic causes of SCA were long QT syndrome genes and RyR2 genes. We summarised the common genes identified in previous SUD and SCA studies in table 3. RYR2 and SCN5A were the most common genes identified, followed by KCNH2, MYPBC3 and KCNQ1. Similarly, several minor LQT syndrome genes and genes involving arrhythmogenic right ventricular cardiomyopathy (ARVC) and cardiomyopathies are also common causes. Patients with cardiomyopathies are known to have risk of SCD, and LVNC and ARVC are notorious to have risk of life-threatening arrhythmia and SCD even before ventricular function deterioration.23 In one large series study of SCD in Denmark, cardiomyopathy is the second most common cause followed by sudden arrhythmia death syndrome in teenagers.22 Therefore, although RYR2 and common LQT syndrome genes are important causes, ARVC and cardiomyopathies genes are also crucial and should be searched aggressively.
For the minor LQT genes, the mother of our patient who had KCNJ2 pathogenic variant (patient 5) had the same mutation and clinically manifested as periodic paralysis and aborted SCA episode during later follow-up. As to the compound variants with SCN5A and Cav 3 (patient 10), we had performed site-directed mutagenesis and coexpression study in cell culture, from which functional correlates from the interaction of these two genes had been noted (not included in this manuscript). These results indicate these minor LQT genes may still have important roles in causing SCA in these young patients.24 This also highlights the important role of whole-exome sequencing, an expanded state-of-the-art panel, to search for the genetic causes of survivors of SCA.
Outcome
Early genetic testing using the state-of-the-art composite genetic panel was associated with long-term outcomes. All patients who received the composite panel were alive at the last follow-up and tended to have better transplant-free survival. In addition, adding the composite genetic panel may increase genetic diagnostic rate, which enhances effective medication use and decreases the ICD shock rate in these survivors of SCA, as shown in figure 1. Such a genetic testing-incorporated approach is very promising but still needs to be confirmed by a large-scale study.
Study limitations
As a retrospective study, we did not recall all patients back for the genetic study. In the past, we have not routinely stored DNA of all survivors of SCA. Therefore, we can only study the nature and genetic causes of survivors of SCA who survived to discharge but not all of the patients with return of spontaneous circulation. For those who did not undergo the genetic test initially at the disease onset, four have died or underwent heart transplantation before we could recruit them for composite panel genetic test, and the other four were lost to follow-up. This may cause selection bias.
Conclusion
The investigation of young unexplained cardiac arrest involves an algorithmic approach, including the clinical history, family history and detailed investigations, which may include pharmacological provocation tests.21 22 The present study shows that the additional use of genetic testing results in improved survival, most likely due to the additional diagnostic value allowing the more precise guidance of protective therapy. Furthermore, a broad genetic panel has a considerably higher yield than a restricted one, which also potentially facilitates effective family screening.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
This study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki, as reflected in a priori approval by the human research committee of our hospital with approval number 201612119RIND. Signed informed consent was obtained from all patients (or their parents if the patients were <18 years of age).
Acknowledgments
The authors thank the laboratory department of medical genetics for their contribution to the study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
S-NC and J-MJJ contributed equally.
Contributors S-NC and J-MJJ conceptualised and designed the study and data collection, drafted the initial manuscript, and reviewed and revised the manuscript. W-CT, N-CL and W-PC designed the data collection instruments, collected data, carried out the initial analyses, and reviewed and revised the manuscript. M-HW conceptualised and designed the study, coordinated and supervised data collection, and critically reviewed and revised the the manuscript. All authors approved the final manuscript as submitted and agreed to be accountable for all aspects of the work.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.