Article Text

Abstract
Objective Early diagnosis and treatment initiation are important factors for successful treatment of mucopolysaccharidosis type I (MPS I). The purpose of this observational study was to assess whether age at diagnosis and time to first treatment for individuals with MPS I have improved over the last 15 years.
Study design Data from the MPS I Registry (NCT00144794) for individuals with attenuated or severe disease who initiated therapy with laronidase enzyme replacement therapy (ERT) and/or hematopoietic stem cell transplantation (HSCT) between 1 January 2003 and 31 December 2017 were included.
Results Data were available for 740 individuals with attenuated (n=291) or severe (n=424) MPS I (unknown n=25). Median age at diagnosis for attenuated disease did not change over time and ranged between 4.5 and 6 years of age while the median duration from diagnosis to first ERT decreased from 5.6 years before/during 2004 to 2.4 months in 2014–2017. For severe MPS I treated with HSCT, median age at diagnosis was less than 1 year and median time to first treatment was less than 3 months throughout the 15-year observation period.
Conclusions Times to diagnosis and HSCT initiation for individuals with severe MPS I were consistent over time. For individuals with attenuated MPS I, the time to ERT initiation after diagnosis has improved substantially in the last 15 years, but median age at diagnosis has not improved. Efforts to improve early diagnosis in attenuated MPS I are needed to ensure that patients receive appropriate treatment at the optimal time.
- metabolic
- monitoring
- multidisciplinary team-care
- therapeutics
Data availability statement
Data may be obtained from a third party and are not publicly available. The data that support the findings of this study can be requested by MPS I Registry participants through a MPS I Registry Data Analyses Request form. The data are not publicly available due to privacy or ethical restrictions. For additional information, please contact rarediseaseregistries@sanofi.com.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
What is already known on this topic?
Mucopolysaccharidosis type I (MPS I) is an inherited lysosomal storage disease where affected individuals experience significant disease burden, disability and premature death.
Early treatment with enzyme replacement therapy and/or haematopoietic stem cell transplantation can reduce/halt disease progression and improve outcomes. However, diagnosis, and consequently the start of therapy, is often delayed, particularly for patients with attenuated phenotypes.
What this study adds?
Our analyses show that while time to diagnosis for attenuated MPS I is still delayed, time to treatment has improved significantly over the 15 years since enzyme replacement therapy was introduced.
Introduction
Mucopolysaccharidosis type I (MPS I) results from deficient α-L-iduronidase, a lysosomal enzyme responsible for glycosaminoglycans (GAGs) dermatan and heparan sulfate metabolism.1 MPS I is a pan-ethnic, autosomal-recessive disease, with an incidence that varies across regions and is estimated as 1/100 000 live births.2 3 Disease phenotypes range from severe (Hurler syndrome) to attenuated (Hurler-Scheie and Scheie syndromes) depending on presence or absence of central nervous system (CNS)/neurocognitive involvement and rate of disease progression.1 4 5 If untreated, MPS I results in significant disease burden and disability, with premature death possible from respiratory and cardiac disease.2
Treatment options include haematopoietic stem cell transplantation (HSCT) for severe disease, and enzyme replacement therapy (ERT) with laronidase (recombinant human α-L-iduronidase; Aldurazyme) for treatment of non-CNS manifestations of MPS I6–9 and in the peritransplant period for severe disease.10 Treatment outcomes are influenced both by disease severity and age at treatment initiation.11 12 Early treatment considerably improves patient outcomes during long-term therapy and is crucial to reduce or halt disease progression before irreversible damage occurs.12–16 However, diagnosis of MPS I is often delayed, particularly for patients with attenuated phenotypes, resulting in delayed introduction of treatment.17–23
Data from the MPS I Registry were analysed to understand trends in diagnosis and treatment in individuals with severe or attenuated MPS I since introduction of ERT and to identify potential factors influencing delays in times to diagnosis and/or treatment (eg, regional differences; disease severity). These data will help us to understand where improvements are still needed to decrease the time to diagnosis and treatment in order to ensure that individuals with MPS I receive the appropriate treatments before significant onset of disease.
Methods
Registry
The MPS I Registry (NCT00144794) is a voluntary, observational global longitudinal database established to capture long-term data to help understand the natural history, standards of care and treatment outcomes of individuals with MPS I.24 Individuals are enrolled in the MPS I Registry by their clinician and data are collected both retrospectively and prospectively as previously described.24 The Registry is overseen and directed by an independent Board of Advisors comprised of physicians who are experts in the care of MPS disorders and is sponsored by Sanofi Genzyme (Cambridge, MA). All participants (patients or caregivers) provide written informed consent for use of anonymised data.
Participants
Observations for individuals in the Registry with either severe or attenuated MPS I were included if MPS I treatment was received between 1 January 2003 and 31 December 2017. For an observation to be included, valid dates for diagnosis and start of treatment were required.
Data management and analysis
Current, primary treatments for analysis included HSCT or ERT (not including peritransplant ERT). For patients with more than one HSCT, first HSCT date was used as start of primary treatment date. Patients with ERT as primary treatment did not receive HSCT. Those for whom HSCT was primary treatment and peritransplant ERT was received, ERT was not counted as primary treatment.
ERT with laronidase became available in 2003, and the MPS I Registry also began enrolling participants in 2003. Data were stratified by MPS I phenotype into year intervals at first primary MPS I therapy. Four intervals were selected for data analysis: in/before 2004 to coincide with initiation of the Registry and laronidase ERT and groupings of 2005–2008, 2009–2013 and after 2013–2017 to have similar time periods covered by each group. Data were also stratified by region where laronidase is approved for commercial use, and presented when sufficient patient numbers for comparisons were available.
All data were analysed as entered by the clinical site. Descriptive statistics include number, percentage, mean, median, SD and IQRs.
Results
Among all participants in the MPS I Registry from January 2003 to December 2017, 740 had data for age at diagnosis and age at treatment. The distribution by region and timing of enrolment for individuals with treatment for MPS I are shown in table 1, with most participants enrolled before 2009. Distribution of participants was evenly divided between Europe (45.8%) and North America (44.5%) with a small percentage of individuals from Latin America (7%) or Asia Pacific (2.7%).
Regional distribution of registry participants with treatment history
Table 2 shows the demographic and clinical characteristics overall and stratified by first primary treatment (ERT or HSCT). Males and females were equally represented. In the entire cohort, 424/740 (57.3%) were categorised with severe disease and 291/740 (39.3%) with attenuated disease, whereas 25/740 (3.4%) did not have a phenotype assigned.
Demographic and clinical characteristics
HSCT treatment trends over time for pretreatment regimens and transplant type and peritransplant ERT are shown in figure 1. The trends generally follow the changes in HSCT recommendations and guidelines that evolved with time and experience. For the most recent time interval, cord blood was the most common source of stem cells (figure 1A) reported in the Registry, and pretransplant conditioning with fludarabine, busulfan and/or antithymocyte globulin/serum or antilymphocyte globulin/serum were the most common regimens. Peritransplant ERT was used pretransplant and/or post-transplant in increasing numbers of patients over time (figure 1B).
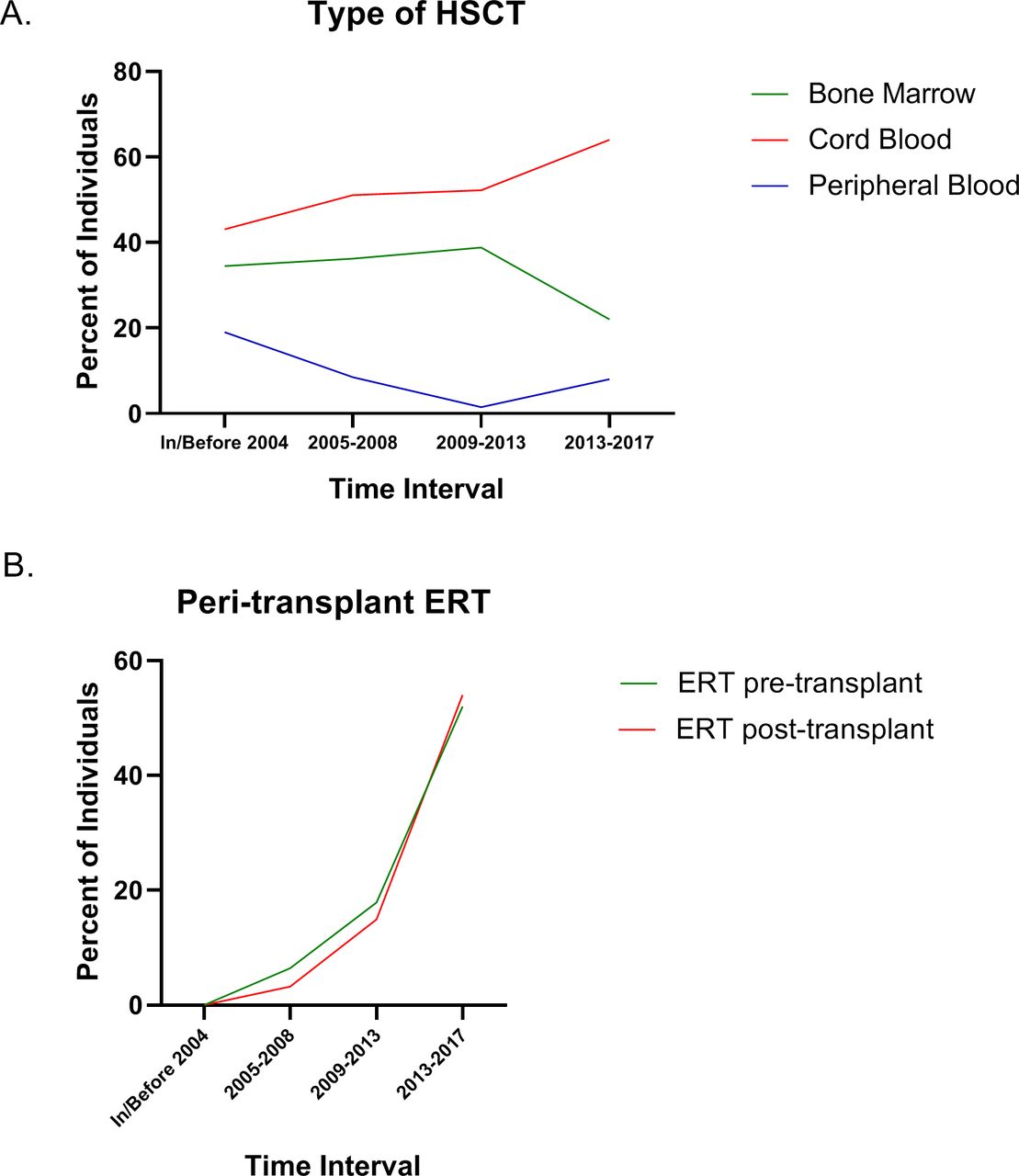
Characteristics of HSCT over time for individuals enrolled in the MPS I Registry. The type of transplant performed showing the source of stem cells is shown in (A). Use of ERT either before or immediately after transplant is shown in (B). ERT, enzyme replacement therapy; HSCT, haematopoietic stem cell transplants; MPS I, mucopolysaccharidosis type I.
Age at MPS I diagnosis
Ages at diagnosis stratified by year interval of first primary therapy and region are shown for individuals with severe or attenuated MPS I for whom ERT was first primary therapy in figure 2 and for HSCT as first primary therapy for individuals with severe MPS I in figure 3.
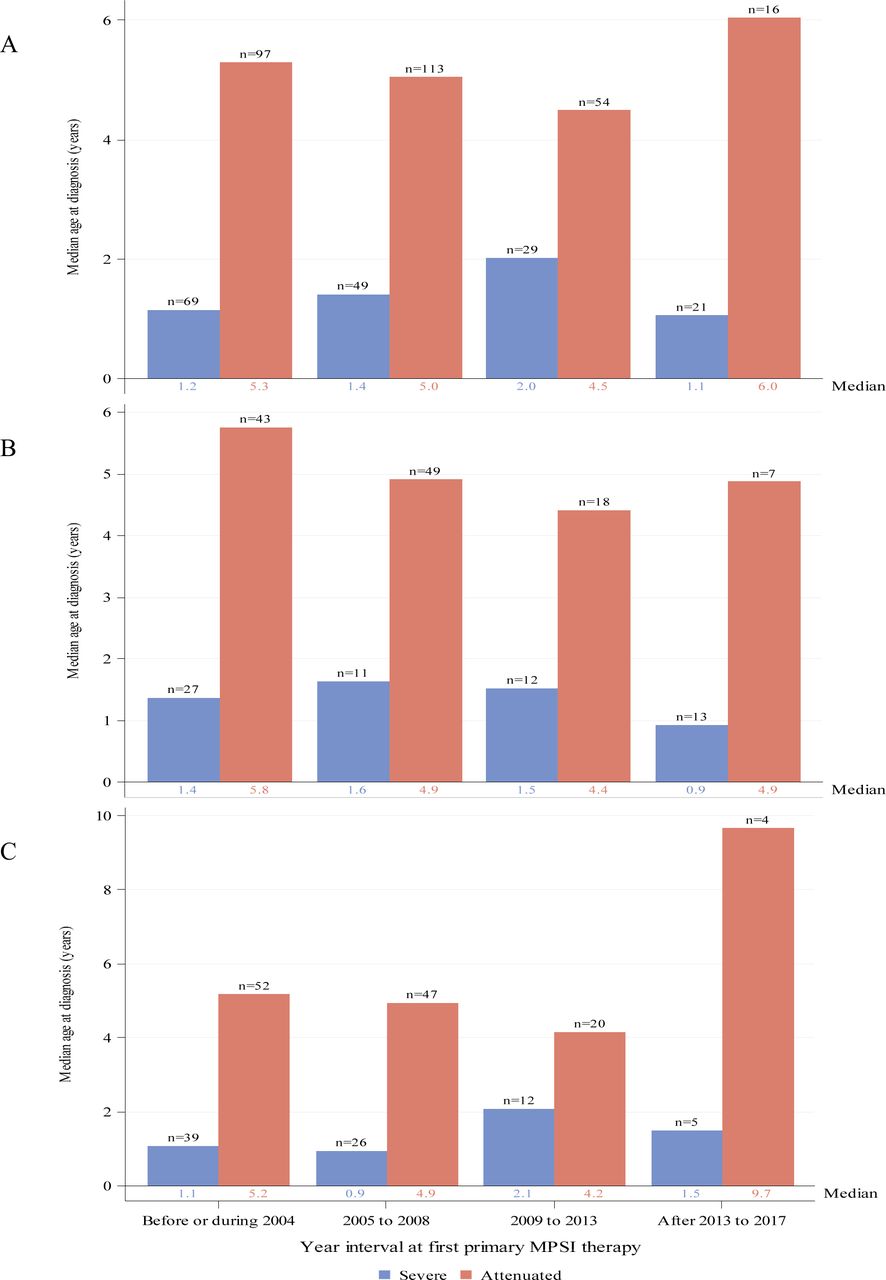
Median age in years at MPS I diagnosis by year interval for individuals with severe (blue) or attenuated (red) MPS I receiving ERT as primary therapy. Global distribution is shown in panel (A). Regional distributions are shown for North America (B) and Europe (C). Individual medians are shown at the base of each bar and individual n’s are shown above the bars. ERT, enzyme replacement therapy; MPS I, mucopolysaccharidosis type I.

Median age in years at MPS I diagnosis by year interval for individuals with severe MPS I receiving HSCT as primary therapy. Global distribution is shown in panel (A). Regional distributions are shown for North America (B) and Europe (C). Individual medians are shown at the base of each bar and individual n’s are shown above the bars. ERT, enzyme replacement therapy; HSCT, haematopoietic stem cell transplants; MPS I, mucopolysaccharidosis type I.
Median ages at diagnosis for individuals with attenuated disease did not change appreciably over time since introduction of ERT and varied between 4.5 and 6 years of age (figure 2A). Among all individuals with severe disease receiving ERT as first primary therapy (these individuals did also not receive HSCT), ages at diagnosis remained fairly stable and varied between 1 and 2 years of age (figure 2A). Medians (IQR) for the intervals before/during 2004, 2005–2008, 2009–2013 and after 2013 to 2017 were 5.3 (3.4, 9.4), 5.0 (3.0, 10.2), 4.5 (3.3, 8.0) and 6.0 (4.2, 21.8) years, respectively, for the attenuated group and 1.2 (0.6, 2.1), 1.4 (0.7, 2.5), 2.0 (0.7, 3.9) and 1.1 (0.7, 2.4) years, respectively, for the severe group. Results were similar for individuals from North America (figure 2B) or Europe (figure 2C).
Median ages at diagnosis for individuals with severe disease receiving HSCT as first primary therapy were 0.8 or 0.9 years for the four time intervals (figure 3A). Median (IQR) for the intervals before/during 2004, 2005–2008, 2009–2013 and after 2013 to 2017 were 0.8 (0.6, 1.1), 0.9 (0.7, 1.2), 0.8 (0.5, 1.1) and 0.9 (0.6, 1.2) years, respectively, and in general, were lower than those for individuals with severe disease receiving ERT as first therapy. Similar results were noted regionally for individuals with severe disease (figure 3B,C).
Time to first primary treatment
Times from diagnosis to first primary treatment stratified by phenotype, year interval of first primary therapy and region are shown for ERT as first primary therapy in figure 4 and for HSCT as first primary therapy in figure 5.

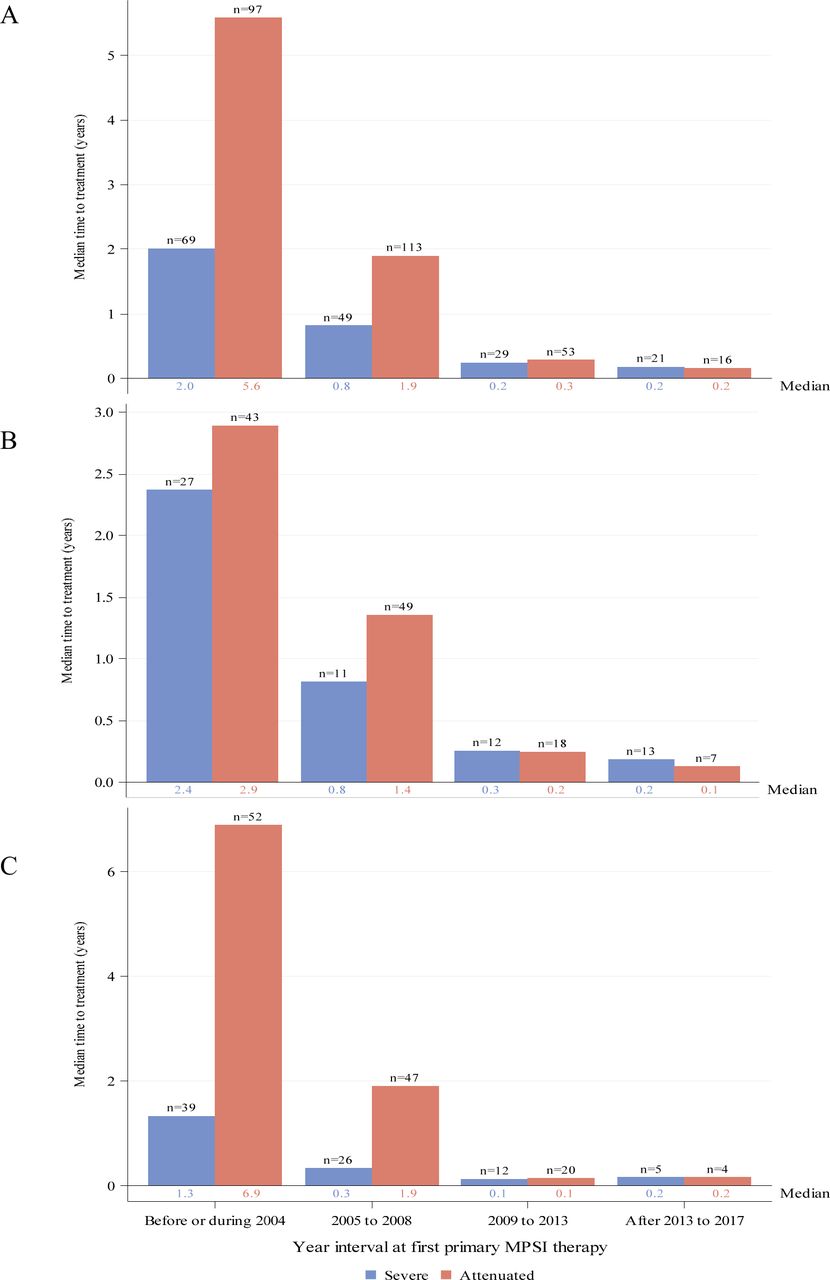
Median duration from diagnosis to first ERT treatment by year interval for individuals with severe (blue) or attenuated (red) MPS I receiving ERT as primary therapy. Global distribution is shown in panel (A). Regional distributions are shown for North America (B) and Europe (C). Individual medians are shown at the base of each bar and individual n’s are shown at top of bars. ERT, enzyme replacement therapy; MPS I, mucopolysaccharidosis type I.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
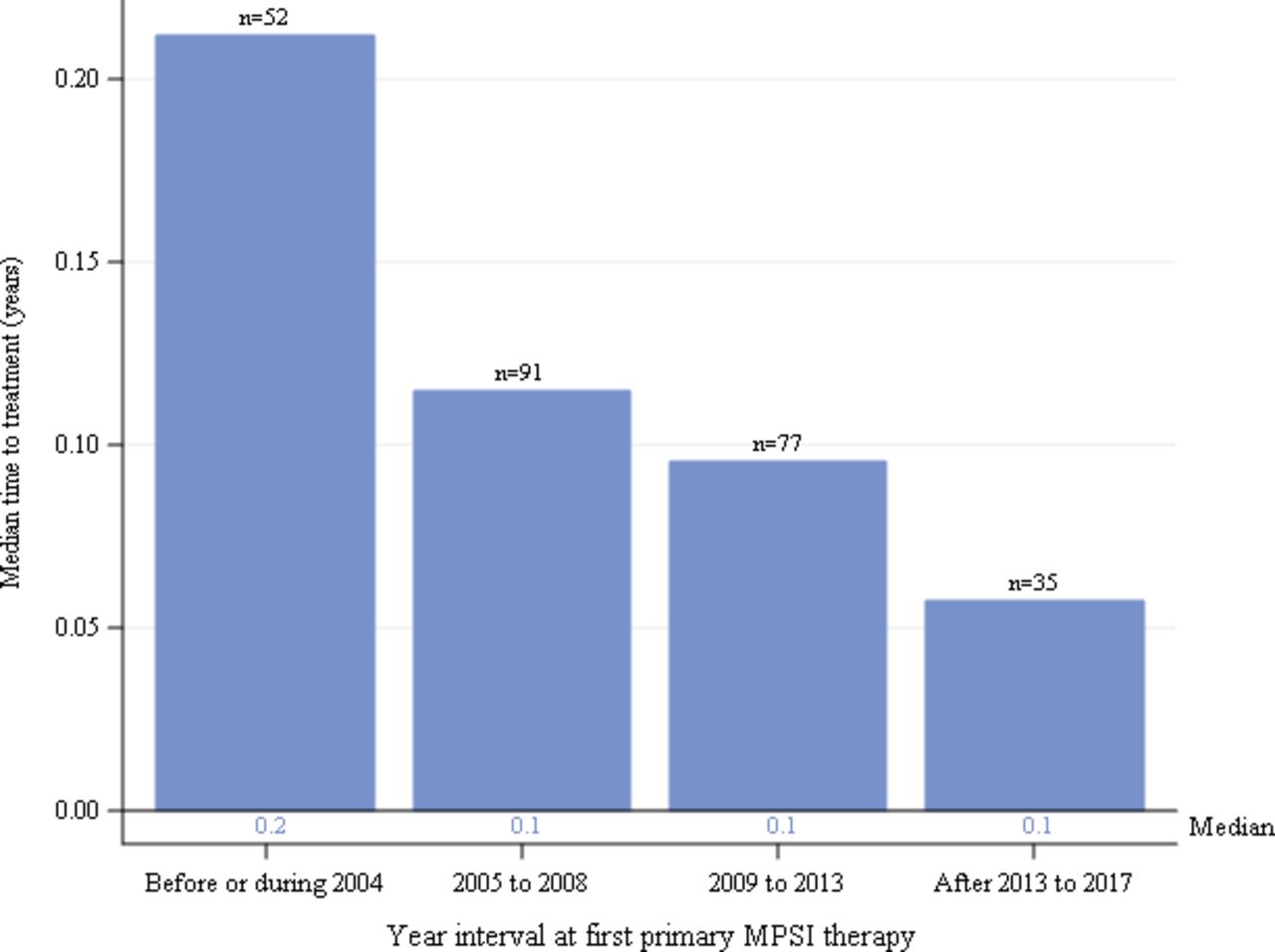
Median duration from diagnosis to first HSCT treatment by year interval for individuals with severe MPS I receiving HSCT as primary therapy. Global distribution is shown. HSCT, haematopoietic stem cell transplants; MPS I, mucopolysaccharidosis type I.
The median durations from diagnosis to first ERT for all individuals with attenuated or severe MPS I decreased over time since introduction of ERT (figure 4A). Medians varied from 5.6 years for individuals with attenuated disease before or during 2004 to 2.4 months in the most recent time interval for both severe and attenuated disease. Medians (IQR) for the intervals before/during 2004, 2005–2008, 2009–2013 and after 2013 to 2017 were 5.6 (1.9, 11.9), 1.9 (0.4, 5.2), 0.2 (0.1, 0.8) and 0.2 (0.1, 0.9) years, respectively, for the attenuated group and 2.0 (0.6, 4.8), 0.8 (0.1, 2.0), 0.2 (0.1, 0.4) and 0.2 (0.1, 0.5) years, respectively, for the severe group. When the data were stratified by region, there was a greater duration from diagnosis to treatment at the earlier time intervals for individuals in Europe (figure 4C) compared with North America (figure 4B). Durations were similar by region in the more recent time intervals.
The median durations from diagnosis to first HSCT for all individuals with severe MPS I were very short for all time intervals (figure 5). Medians (IQR) were 0.2 (0.1, 0.3), 0.1 (0.1, 0.3), 0.1 (0, 0.2) and 0.1 (0, 0.1) years for intervals before/during 2004, 2005–2008, 2009–2013 and after 2013 to 2017, respectively. When stratified by North American and European regions (not shown), results were similar to the global data.
Discussion
First signs and symptoms in individuals with MPS I vary in type, severity and age of onset,18 24 25 which can lead to significant diagnostic delays due to the non-specific nature of symptoms that are suggestive of other diseases. This is particularly true for individuals with attenuated MPS I.17–19 24 In a study of the diagnostic history of 60 individuals with attenuated MPS I from the USA, Europe and Latin America, participants reported that, on average, there was a 3-year delay between first physician visit and diagnosis for attenuated MPS I, and 20% had delays of 5 years or longer.20 These are similar to regional surveys of MPS I patients/caregivers conducted in the USA, Latin America and the Netherlands.21–23 Referrals to rheumatologists, orthopaedists, pulmonologists and gastroenterologists are common once children with MPS I present to paediatricians.26 Regardless of disease severity or initial symptoms, a mean of five specialists were consulted before receiving a correct diagnosis, highlighting the continued need for awareness education within the medical community.20
Our analyses involve a large dataset of diagnosis and treatment trends over time for individuals with MPS I. Time intervals for analysis began with approval of laronidase ERT by the FDA and EMA in 2003. We found that delays in diagnosis for individuals with attenuated MPS I have not improved over time. For patients not identified by newborn screening (NBS), algorithms to raise early clinical suspicion of MPS I based on the range of key signs and symptoms,26–29 are available for clinicians so that full diagnostic assessment30 of enzyme activity, substrate levels (ie, GAGs) and molecular analyses is conducted for appropriate patients.30 It is worth noting that despite being a simple test, measurement of urinary GAGs is not available in all regions, and shipment of urine samples to reference laboratories may be difficult to arrange. In addition, urinary GAG levels may not be informative in young infants due to lack of standardisation of age-related reference ranges,31 The use of dried blood (DBS) for measurement of GAGs may provide useful information before another sample is requested.32
It is hoped that educational initiatives in combination with evolving NBS programmes worldwide,30 31 33–36 will decrease time to MPS I diagnosis and treatment. NBS for MPS I have used either tandem mass spectrometry or digital microfluidic to measure IDUA enzyme activity in DBS.34 36 Follow-up of a positive NBS result (deficient IDUA activity) should include confirmatory IDUA enzyme activity in leucocytes, serum or plasma, urine GAG analysis and molecular analysis.31 The presence of IDUA pseudodeficiency alleles (benign variants) complicates NBS for MPS I since the pseudodeficiency alleles are more common than pathogenic IDUA variants.37 Pseudodeficiency is decreased IDUA activity with no evidence of GAG storage. Second-tier testing measuring either GAG levels or sequencing of the IDUA gene using the original DBS has been used to reduce the NBS false-positive rate and the follow-up burden.32 36–38 The MPS I phenotype can be predicted based on genotyping in the majority of cases,39 and early treatment initiated based on predicted phenotype for severe MPS I (HSCT with or without peritransplant ERT) or intravenous ERT for attenuated phenotypes.31 However, infants with abnormal NBS whose phenotypes cannot be positively predicted (ie, IDUA variants of unknown significance) require careful clinical and biochemical monitoring in order to identify appropriate treatment.31 35 36
In general, individuals with severe MPS I are diagnosed at a younger age than those with attenuated disease, in part due to the more pronounced somatic symptoms. Our results mirror previous reports of MPS I Registry data,17 showing that this trend has not changed with time. Early diagnosis of severe MPS I is important since HSCT is indicated before 2 years of age, and early treatment is associated with better outcomes.14 16 40 MPS I management decisions following positive NBS depend on the integration of biochemical, molecular and clinical assessments.30 31 In the case of a patient identified with severe MPS I based on the presence of two pathogenic IDUA variants that predict this phenotype, the patient is referred for HSCT and potentially early peri-HSCT ERT.31 In accordance with current guidelines, HSCT should be ideally performed before 1 year of age, and typically not after 2.5 years, although the upper age limits vary by geographical region.40 41 A 10 year follow-up study showed that peritransplant ERT is associated with good outcomes in patients with severe MPS I.10
Interestingly, for individuals in the MPS I Registry with severe MPS I for whom ERT was the primary therapy, age at diagnosis was older than those with severe MPS I receiving HSCT as primary therapy by up to 1.1 years depending on treatment interval. This may reflect differences in onset of key somatic symptoms in some patients that are hallmarks of severe MPS I and drive diagnosis or differences in regional access to healthcare. We should also consider that patients diagnosed after the age of 2.0 years may not have been eligible for HSCT according to treatment guidelines, driving the patients to ERT monotherapy.42 HSCT is not always the primary treatment option for patients with severe MPS I. In some countries, the logistical and clinical challenges associated with HSCT remain high and families may decide not to take the risk. In addition to age restrictions, there are individuals with severe MPS I for whom HSCT is not a treatment option due to compromised physical state or lack of a suitable donor. Thus, patients with severe MPS I may still receive only ERT, although outcomes with ERT monotherapy in patients with severe MPS I are inferior to those obtained with early HSCT.42
While multiple studies have investigated delays between symptom onset and MPS I diagnosis, there is much less known regarding time between diagnosis and treatment onset. The duration from diagnosis to first treatment with ERT has improved substantially over time since introduction of laronidase ERT in 2003, from a median of over 5 years to less than 2 months. Early treatment with ERT has been shown to considerably improve patient outcomes during long-term therapy.12 13 15 We anticipate that NBS programmes will result in decreased times to diagnosis and treatment with implementation and evolution of best management practices.31 35 42
Strengths and weaknesses of study
As with all observational studies, there exists the potential for ascertainment and reporting biases due in part to the voluntary and observational nature of the MPS I Registry, which may impact the generalisability of the findings. The analyses included registry participants that subsequently died after data were captured on age at diagnosis and time to primary treatment. While a potential for bias is introduced by including patients that died who ultimately may have had severe disease, the bias would likely result in shorter times to diagnosis. The limitations of this analysis include the small numbers of individuals due to the nature of rare diseases, as well as limited data available in some regions. In particular, the smaller number of individuals from Latin America and Asia limit the comparison of these regions’ trends with those for North America and Europe and make it difficult to compare factors such as healthcare system practices that may affect age at diagnosis and time to treatment. Specifically, there are significant differences in the healthcare practices of individual countries with respect to reimbursement of ERT and HSCT that may impact time to treatment, as well as challenges that patients and their families face in travelling to specialty centres for ERT infusions and/or transplants.
Conclusion
Early diagnosis is crucial for the best therapeutic outcomes with both ERT and HSCT to reduce disease progression before irreversible organ and tissue damage occur. The present study demonstrates that diagnosis of MPS I is delayed. However, time to treatment initiation, once individuals have been diagnosed, has improved substantially in the last 15 years for patients with either severe or attenuated MPS I. Efforts to improve early diagnosis are needed to ensure that patients receive appropriate treatment at the optimal time. These data illustrate that across regions, improvement in MPS I awareness is still needed.
Data availability statement
Data may be obtained from a third party and are not publicly available. The data that support the findings of this study can be requested by MPS I Registry participants through a MPS I Registry Data Analyses Request form. The data are not publicly available due to privacy or ethical restrictions. For additional information, please contact rarediseaseregistries@sanofi.com.
Ethics statements
References
Footnotes
Contributors RG contributed to the conception and design of the manuscript, analysis and interpretation of the data, drafted and revised the manuscript, approved the final version and is accountable for all aspects of the manuscript. NM contributed to interpretation of the data, revised the manuscript, approved the final version and is accountable for all aspects of the manuscript. MD contributed to interpretation of the data and revision of the manuscript, approved the final version and is accountable for all aspects of the manuscript. HK contributed to the conception and design of the manuscript, analysis and interpretation of the data, drafted and revised the manuscript, approved the final version and is accountable for all aspects of the manuscript. JM contributed to the conception and design of the manuscript, analysis and interpretation of the data, drafted and revised the manuscript, approved the final version and is accountable for all aspects of the manuscript.
Funding The study was funded by Sanofi Genzyme, Cambridge, Massachusetts, USA. Patrice C Ferriola, PhD, provided medical writing and editing assistance and was funded by Sanofi Genzyme.
Competing interests RG has received consultant/speaker honoraria/fees and/or served on advisory boards for Actelion, Amicus, Biomarin, Inventiva, RegenxBio, Sanofi Genzyme, Sobi, Takeda and Ultragenyx, conducts contracted research for BioMarin, JCR, Lysogene, RegenxBio, Sanofi Genzyme, Takeda, and Ultragenyx and is a member of the International Board of Advisors of the MPS I Registry. NM is a consultant for Biomarin, Sanofi Genzyme, Lysogene, Shire and Sobi, has received grants/research support from Amicus, Biomarin, Sanofi Genzyme and Shire and has received honoraria and/or travel grants from Actelion, Amicus, BioMarin, Chiesi Farmaceutici S.p.A., Sanofi Genzyme and Shire. HK was an employee of Sanofi Genzyme at the time of the study.MD has received consultant/speaker honoraria and/or travel grants from Biomarin, Sanofi Genzyme, Regenxbio, Bluebird Bio, Ultragenyx. JM received consultant/speaker honoraria/fees and served on advisory boards for BioMarin, Denali, Eloxx, RegenxBio, Sangamo, Sanofi Genzyme and Shire, is a PI for a Phase I/II and Phase II/III intrathecal enzyme replacement clinical trials for MPS II and a Phase I/II gene editing clinical trial for MPS II and is a member of the International Board of Advisors of the MPS I Registry.
Provenance and peer review Not commissioned; externally peer reviewed.