Article Text

Abstract
Objectives To report investigations performed in children with progressive neurodegenerative diseases reported to this UK study.
Design Since 1997 paediatric surveillance for variant Creutzfeldt-Jakob disease (vCJD) has been performed by identifying children aged less than 16 years with progressive intellectual and neurological deterioration (PIND) and searching for vCJD among them.
Setting The PIND Study obtains case details from paediatricians who notify via the British Paediatric Surveillance Unit.
Participants Between May 1997 and October 2017, a total of 2050 cases meeting PIND criteria had been notified and investigated.
Results Six children had vCJD. 1819 children had other diagnoses, made in 12 cases by antemortem brain biopsy and in 15 by postmortem investigations. 225 children were undiagnosed: only 3 had antemortem brain biopsies and only 14 of the 108 who died were known to have had autopsies; postmortem neuropathological studies were carried out in just 10% (11/108) and only two had prion protein staining of brain tissue. Of the undiagnosed cases 43% were known to come from Asian British families.
Conclusions Most of the notified children had a diagnosis other than vCJD to explain their neurological deterioration. None of the undiagnosed cases had the clinical phenotype of vCJD but brain tissue was rarely studied to exclude vCJD. Clinical surveillance via the PIND Study remains the only practical means of searching for vCJD in UK children.
- epidemiology
- neurology
- neuropathology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
What is already known on this topic?
Since 1997, the PIND Study has identified six UK children with variant Creutzfeldt-Jakob disease (vCJD).
In 2017, vCJD was reported in a man who was methionine/valine (MV) heterozygous at PRNP codon 129.
This novel finding raises the possibility of a second wave of vCJD cases in UK individuals with the MV genotype.
What this study adds?
Most of the Study children had a diagnosis other than vCJD to explain their neurological deterioration but some of them remained undiagnosed even after death.
Of the 225 undiagnosed PIND children, only 10% of those who died had postmortem neuropathological studies; only 2 had prion protein staining of brain tissue.
The low autopsy rate in undiagnosed children emphasises the need to obtain antemortem or postmortem samples of blood and other relevant tissues for later study.
Introduction
The first cases of variant Creutzfeldt-Jakob disease (vCJD) in young adults were reported by the National Creutzfeldt-Jakob Disease Research and Surveillance Unit (NCJDRSU) in 1996.1 Until 2016, all the 177 definite and probable cases of vCJD2 had occurred in patients who were methionine homozygous (MM) at codon 129 of the prion protein gene (PRNP). However, in January 2017, it was reported that vCJD had been confirmed in a man who was methionine/valine (MV) heterozygous at PRNP codon 129.3 In the general population of the UK about 44% have the MM genotype and 45% are MV,4 which raises the possibility of a second wave of vCJD cases in those with the MV genotype. In addition, there are concerns about surgical5 and bloodborne transmission,6 so there is a need for continuing national surveillance for vCJD.
When vCJD was reported in adults it raised the possibility that it might occur in children and it was therefore necessary to search systematically for cases of vCJD among the many neurodegenerative disorders of childhood. In 1997, the PIND Study commenced via the British Paediatric Surveillance Unit (BPSU). Paediatricians in the UK are asked to notify all patients aged less than 16 years with ‘progressive intellectual and neurological deterioration’ (PIND) because this group of children is likely to include any cases of vCJD.
Most of the children notified to the PIND Study have an underlying diagnosis other than vCJD to explain their deterioration. However, vCJD could be hidden among undiagnosed cases. This paper considers the public health issues raised by this undiagnosed group, including an analysis of any differences according to patient ethnicity as there are relatively large numbers of PIND cases in certain ethnic groups.7
Methods
The BPSU has studied rare childhood disorders since 1986.8 All UK consultant paediatricians are requested to notify children with conditions under surveillance using a monthly online system. Currently 3580 paediatricians are involved and over 90% reply. The PIND Study has used the BPSU system since 1997,9 asking for notifications of all children meeting the case definition for PIND shown in box 1 (see the protocol in the BPSU section of the Royal College of Paediatrics and Child Health website10).
Case definition—progressive intellectual and neurological deterioration
Any child (under 16 years of age at onset of symptoms) who fulfils all of the following three criteria:
Progressive deterioration for more than 3 months.
Loss of already attained intellectual or developmental abilities.
Development of abnormal neurological signs.
Excluding:
Static intellectual loss, for example, after encephalitis, head injury or near drowning.
Including:
Children who meet the case definition, even if specific neurological diagnoses have been made.
Metabolic disorders leading to neurological deterioration.
Seizure disorders if associated with progressive deterioration.
Children who have been diagnosed as having neurodegenerative disorders who have not yet developed symptoms.
Reporting restricted to cases seen in the last month but including those whose condition began earlier (ie, including ‘old cases’ of children in follow-up if seen in that month).
Clinical information is obtained via a questionnaire completed by the notifying paediatrician, via telephone interview or by site visit; there is no contact with the patients or their families. The study records information about presenting symptoms and signs and follows progress after presentation.
Classification of ethnicity is that used by the then Communicable Disease Surveillance Centre, Colindale, London, UK, when the PIND Study commenced in 1997. The categories are: White, Black, Indian, Pakistani, Bangladeshi, Chinese and Other. Some clinicians reported that their patients were ‘Asian unspecified’. For the current analysis the categories Indian, Pakistani, Bangladeshi, Chinese and ‘Asian unspecified’ were combined under the heading ‘Asian British’ for comparison with the 2011 census of England and Wales where these groups were combined as ‘Asian/Asian British’.11 The PIND questionnaire asks if the parents of notified children are consanguineous, without defining the term, but the details were obtained and only close blood relatives (ie, cousins) were included in this group.
The clinical data are anonymised by the PIND Study team and are then reviewed by an independent PIND Expert Group to confirm and classify diagnoses. The Group consists of paediatric neurologists, specialists in paediatric metabolic disease and representatives from the NCJDRSU. If vCJD is suspected it is suggested that the local paediatrician obtains consent to refer the child to the NCJDRSU in Edinburgh. Otherwise, the notified children are followed up by the PIND team via the local clinician until: (A) a definite diagnosis is made, (B) no further investigations are planned, or (C) the child dies. These children are often investigated by several national or international experts. Some remain undiagnosed despite all investigations; they are not systematically followed up by the PIND Study, however the notifying paediatricians are asked to inform the Study if a diagnosis is subsequently made.
For this paper, an autopsy is defined as: ‘dissection and systematic examination of a body after death in order to determine the cause of death or the presence of disease processes’. The term ‘post-mortem pathological examination’ is used to include full autopsies as well as more limited postmortem examinations or biopsies of one or more tissues after death.
Consent for research was granted by Addenbrooke’s Cambridgeshire 2 Research Ethics Committee (Ref: 97/010); Patient Information Advisory Group/BPSU 2-10(c)(2005), National Information Governance Board, and Confidentiality Advisory Group under Section 251 of the National Health Service (NHS) Act 2006. Sponsored by Cambridge University Hospitals NHS Foundation Trust, R&D reference A05001. Registered on the National Research Register (ID N0287023055). UKCRN ID 6636.
Results
Between May 1997 and October 2017, a total of 4244 children had been notified to the PIND Study. Of these, 1933 were found not to meet the PIND criteria and 261 were either still under investigation or clinical details were pending. The other 2050 cases met the PIND criteria and had been fully investigated; 6 had vCJD (identified between 1997 and 2000)9 12 and 1819 (89% of the PIND cases) had a diagnosis other than vCJD to explain their deterioration. In the remaining 225 children (11% of the PIND cases) investigations had been negative or they had died undiagnosed.
The 1819 diagnosed cases
In the diagnosed group 977 were male, 842 were female; the ethnic distribution is shown in figure 1. The proportion of Asian British families in the diagnosed group was 29% (483/1683 for whom ethnicity was known). The rate of consanguinity in the diagnosed group was 32% (461/1440 for whom consanguinity was known). The 483 Asian British children comprised: 327 Pakistani, 43 Indian, 43 Bangladeshi, 5 Chinese and 65 Asian unspecified. The rate of consanguinity in the families of Pakistani origin was 85% (279/327). In the whole diagnosed group there were more than 190 different disorders; figure 2 shows the 10 disorders identified most frequently.
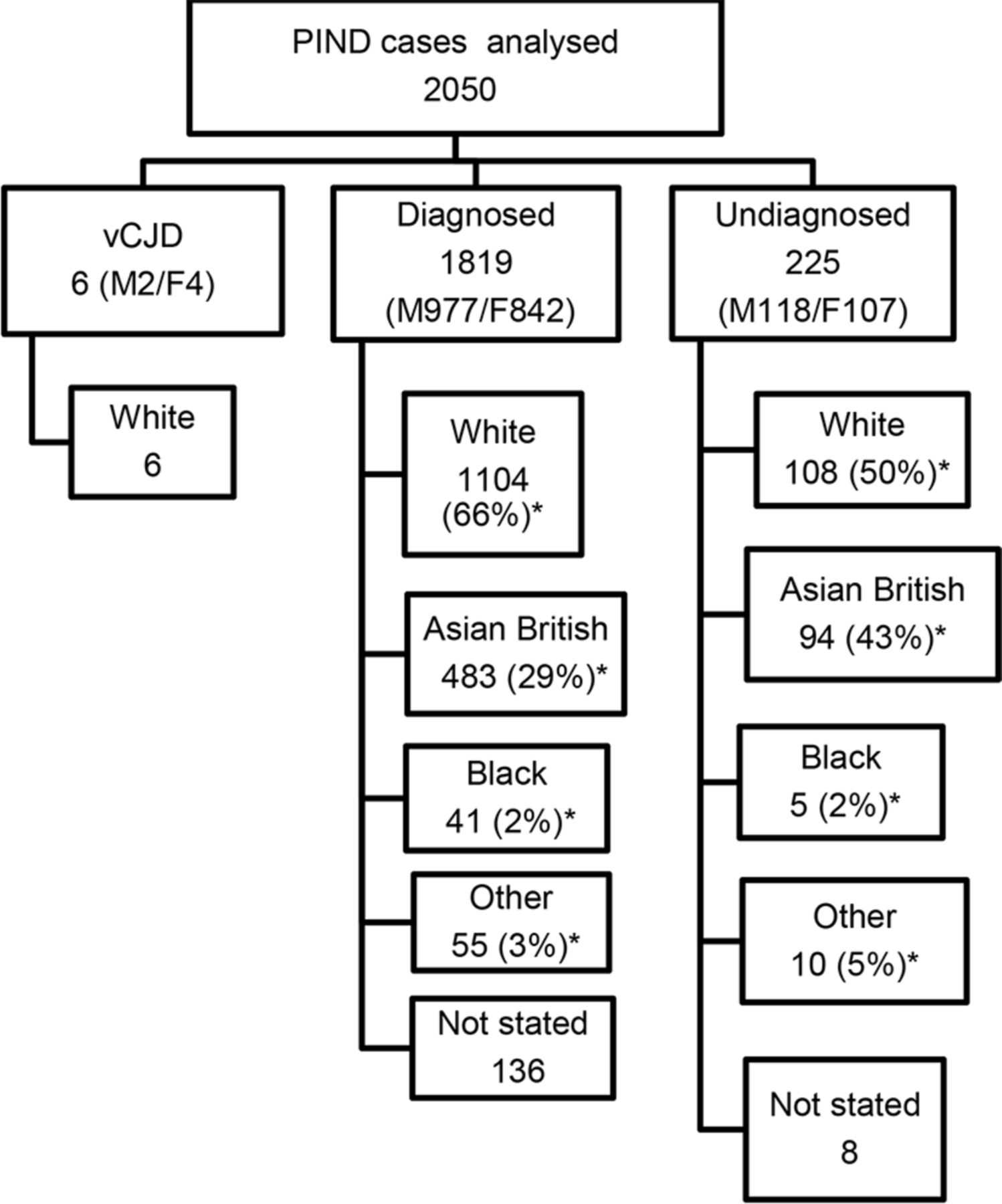
Flow chart showing the ethnicity of the 2050 cases that met the PIND criteria and had been fully investigated. *Proportion for whom ethnicity was known. F, female; M, male; PIND, progressive intellectual and neurological deterioration; vCJD, variant Creutzfeldt-Jakob disease.

{kind=link}
{kind=link}
The 10 most common diagnoses of more than 190 different disorders in the 1819 diagnosed children with progressive intellectual and neurological deterioration identified between May 1997 and October 2017. ALD, adrenoleukodystrophy; Krabbe, Krabbe disease; MLD, metachromatic leukodystrophy; MPS III, mucopolysaccharidosis type III; NCL, neuronal ceroid lipofuscinosis; NP-C, Niemann-Pick disease type C; Rett, Rett syndrome; Sandhoff, Sandhoff disease; Tay-Sachs, Tay-Sachs disease.
In 13 cases a brain biopsy had been performed during life and 12 helped to make the diagnosis. Of the diagnosed children 241 were reported to have died; information about postmortem examinations was available for 109. Twenty-four of 109 (22%) had postmortem examinations of whom 20 had postmortem neuropathology (17 full autopsies, 1 coroner’s case without neuropathology, 3 limited to brain and spinal cord pathology, 1 limited to biopsies of skin, muscle and liver, 1 liver biopsy only and 1 muscle biopsy only). Of the 24 postmortem studies 15 helped to make the diagnosis and nine did not. Thus, the diagnosis was made without information from brain biopsies or postmortem examinations in 1819 − (12+15)=1792 cases (99%). The percentages known to have had postmortems by ethnic group were: White 21/1104=2%, Pakistani 2/327=1% and Indian 1/43=2%. The last reported diagnostic postmortem was in 2007.
The 225 undiagnosed cases
In the undiagnosed group 118 were male, 107 were female; the ethnic distribution is shown in figure 1. There was a high rate of consanguinity—52% of those for whom the data were available (100/194). The proportion of Asian British families in the undiagnosed group was 43% (94 of 217 for whom ethnicity was known). The 94 Asian British children comprised: 72 Pakistani, 7 Indian, 4 Bangladeshi and 11 Asian unspecified. The rate of consanguinity in the families of Pakistani origin was 89% (64/72), compared with 7% (8/108) in the White families.
The 225 undiagnosed cases had usually been very thoroughly investigated with, where appropriate, metabolic tests, neurophysiological studies, brain scans, karyotyping, DNA studies, skin, muscle and rectal biopsies as shown in table 1.
Specialised investigations reported in the 225 undiagnosed cases during life, in addition to extensive biochemical studies of blood and urine
CT brain scans were performed in 65/223 (29%) (2: information not available) and at least one MRI brain scan was performed in 184/224 (82%) (1: information not available). The principal abnormalities found on these scans were heterogeneous (see table 2).
Principal abnormalities reported on CT and MRI brain scans in the 225 undiagnosed cases
It was reassuring that none of the MRI scans were reported to have the pulvinar sign associated with vCJD.13 MRI brain scans were performed in 73/94 (78%) of the Asian British children compared with 92/108 (85%) of the White children, suggesting that Asian British children were being as thoroughly investigated as White children because MRI scans are not easily performed in this age group.
In three undiagnosed children a brain biopsy had been performed during life. In 123 cases the child’s death had been reported. Postmortem pathological examinations had been carried out in 14 (13%) of the 108 cases for whom information was available: 9 had a full autopsy and 5 had partial examinations (in 1 the brain was not examined, in 2 just the brain was examined, in 1 postmortem liver and muscle biopsies were taken and 1 had a postmortem liver biopsy). Postmortem neuropathological examination was carried out in 11 cases, in two of these brain tissue was sent to the NCJDRSU where staining for abnormal prion protein was negative. In another case, the neuropathology was reviewed by the NCJDRSU and was not that of vCJD. In the ethnic groups the percentages that had postmortems were: White 8/108=7%, Pakistani 4/72=6% and Indian 2/7=29%.
Children were not routinely followed by the Study after being allocated to the undiagnosed group. Therefore, in October 2017, the National Health Service Spine Portal was used to discover how many of the 225 undiagnosed cases had died by that time: the total number was 161, which is 38 cases more than had been reported by the notifying paediatricians between May 1997 and October 2017.
Discussion
Principal findings
Postmortems and diagnosis
The diagnosis was made without brain biopsy or postmortem neuropathology in 99% of the whole diagnosed group of children with PIND. Only 24 children in the diagnosed group were known to have had postmortem examinations. However, these did help to make the diagnosis in 15 cases. It is therefore a concern that only 14 of the 108 undiagnosed children who died were known to have had postmortem studies and in three of these the brain was not examined.
Falling autopsy rates have been noted in adults and children.14–16 Burton and Underwood14 reported that the overall autopsy rate in the UK in 1979 was 42.7%, falling to 15.3% in 2001. In 2015, Turnbull et al17 found that the mean autopsy rate in the UK was 0.69%: hospital autopsy rates in children’s hospital NHS trusts ranged from 0% to 21%. A recent systematic review18 of postmortem examinations in perinatal and paediatric settings stated, ‘…PM examinations result in clinically significant findings in 22%–76% of cases…’
Possible hidden vCJD
It is reassuring that the majority of notified UK children who met the PIND criteria had a diagnosis other than vCJD to explain their deterioration. However, 225 of them had no diagnosis—could some of them have vCJD? A previous short report by the PIND Study19 provided reassurance about this and the current data reinforce the view that there is no evidence of hidden vCJD in undiagnosed children with PIND; none fulfilled the sensitive and specific diagnostic criteria for ‘probable’ vCJD (see table 3).20
Current diagnostic criteria for vCJD as reported by Heath et al20 in 2010
However, it is a concern that prion protein staining of brain tissue was carried out in just two of the undiagnosed cases as this is the only way to completely exclude the diagnosis of vCJD.21 Blood and urine tests for diagnosing vCJD have been developed, but are not sufficiently validated for routine clinical use.22–24
Ethnicity and consanguinity
The PIND Study has reported that the greatest numbers of cases come from areas where there are relatively large populations of Pakistani origin.7 These new data confirm that a large proportion of Study children are Asian British (29% in the diagnosed group and 43% in the undiagnosed group compared with 7.5% in the general population of England and Wales in 201111). There were high rates of consanguinity—in the families of undiagnosed children of Pakistani origin with PIND it was 89%: possibly they have inherited neurodegenerative diseases that have not yet been characterised. These diseases may become evident as new diagnostic investigations are available.
Strengths and weaknesses of the PIND Study
The Study uses the well-established health surveillance mechanism of the BPSU.8 The PIND Study Expert Group includes representatives from the NCJDRSU and provides an independent opinion about the diagnosis in every reported case. There is multisource ascertainment and the reporting is not restricted to cases admitted to hospital. The Study has confirmed the diagnosis of more than 190 known neurological disorders and has published case series of the diagnosed groups.25–28
The BPSU relies on the voluntary support of consultant paediatricians: it does not provide complete ascertainment of cases and it is not always possible to obtain all the relevant data. When PIND cases are diagnosed they are not systematically followed up by the Study and follow-up has to be discontinued eventually in undiagnosed cases.
UK surveillance for prion diseases is carried out by the NCJDRSU, the team that first identified this new disease.1 The NCJDRSU relies on a network of neurologists, psychiatrists, neuropathologists and other clinical specialists who report adults with suspected vCJD. Because vCJD cases could be hidden among the multitude of rare neurodegenerative diseases of childhood a system of direct referral of suspected vCJD cases would not be reliable. The PIND Study therefore has to work via local paediatricians and other specialists to screen all the children in the UK with PIND.
Policy implications and future research
There has been increasing use of minimally invasive postmortem investigations in infants and children. Postmortem MRI can provide useful diagnostic information29–31 and may identify significant neuropathology in infants and children.32 Storage of blood and other relevant tissues for metabolic and/or genetic studies is very important; several PIND Study children have been diagnosed via the UK and Ireland Deciphering Developmental Disorders Study.33 Some children with undiagnosed PIND might be eligible for inclusion in the 100 000 Genomes Project.34 Because of the recent advances in genetics undiagnosed children should be referred for an up-to-date opinion from a clinical geneticist.
The recent identification of the first patient with vCJD who was MV heterozygous at PRNP codon 129 reinforces the need for continued vCJD surveillance,3 particularly as a study of archived appendix samples from UK hospitals published in 2013 indicated that approximately 1 in 2000 of the UK population is carrying abnormal prion protein in the gastrointestinal tract.35
Should more children with neurodegenerative diseases have autopsies? In this study, an underlying diagnosis had been made in the majority of children in whom investigations of their neurodegenerative diseases had been completed; in 99% of these 1819 cases the diagnosis was reached before death without the need for brain biopsies or autopsies. Thus, the PIND Study does not provide evidence that autopsies should be performed more frequently in most children with neurodegenerative diseases meeting the Study criteria. However, if such children have been carefully investigated before death and are still undiagnosed there is a case for recommending postmortem studies. There are different considerations when trying to confirm or exclude vCJD, as this can only be done with certainty by neuropathological study. If postmortem studies of brain tissue were always performed after children died of undiagnosed neurodegenerative diseases then vCJD would be detected and there would be no need for the PIND Study. However, the undiagnosed children in the Study rarely underwent antemortem or postmortem pathological examination of brain tissue and this seems unlikely to change. Therefore, in the absence of an alternative validated vCJD screening test the PIND Study remains the only practical means of performing systematic surveillance for vCJD in UK children.
Acknowledgments
Many thanks to the paediatricians who notify cases, to Mr Richard Lynn and Mr Jacob Avis of the BPSU and to the PIND Expert Group: PB, CdS, PG, MK, JL, RM, SP, MP, RR, EW, RW, Dr John Wilson and SZ.
References
Footnotes
Funding This report is based on independent research commissioned and funded by the National Institute for Health Research (NIHR) Policy Research Programme (’To Undertake Prospective Multisource Surveillance for all Cases of Progressive Intellectual and Neurological Deterioration Occurring in Children in the UK' PR-ST-1216-10001).
Disclaimer The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the NIHR, the Department of Health, arm’s length bodies or other government departments.
Competing interests None declared.
Patient consent Not required.
Ethics approval NRES Committee East of England Cambridge Central.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data.