Article Text

Statistics from Altmetric.com
- BMI, body mass index
- BT, biotinylated tyramine
- DAB, 3,3′-diaminobenzidine
- ERG, electroretinogram
- HL, hearing loss
- MTI, myringotomy tube insertion
- NHS, normal horse serum
- PBS, phosphate buffered saline
- PTA, pure tone audiometry
- ROM, relapsing otitis media
- RUTI, recurrent upper respiratory tract infections
- RP, retinitis pigmentosa
- XLRP, X linked recessive RP
Retinitis pigmentosa (RP) is the most common form of hereditary retinal degeneration, affecting approximately 1 in 3500 individuals.1,2 Classical RP is characterised by progressive night blindness and constriction of the peripheral visual fields, ultimately causing deterioration of the central vision in many patients. These symptoms are accompanied by ophthalmoscopically detectable degenerative and pigmentary changes of the retinal tissue and by reductions of the electrical retinal response to flashes of light using an electroretinogram (ERG). ERG abnormalities are typically present before any detectable retinal change becomes visible to clinical examination. RP can be transmitted by all inheritance modes, with X linked recessive RP (XLRP) accounting for 10–20% of genetically identifiable cases and being reportedly among the most severe forms.1,2 To date, two of the genes responsible for XLRP have been cloned: RP23 and the retinitis pigmentosa GTPase regulator, RPGR.4,5RPGR accounts for the majority of XLRP.6,7
RP or RP-like retinopathies can also be part of syndromic conditions—that is, associated with extra-ocular manifestations. The most common are Bardet-Biedl and Usher syndromes.1,8–11 Usher syndrome is characterised by the association of RP with sensorineural hearing loss of variable severity, and has three broad clinical phenotypes, with types I and II being the most common.9,11 Type I is characterised by profound congenital non-progressive hearing loss, marked speech impediment, and vestibular dysfunction. The hearing loss of Usher syndrome type II is typically mild to moderate, limited to high frequencies, non-progressive in nature, associated with speech abnormalities commensurate to the hearing defect, and with normal or minimally abnormal vestibular function. Usher syndrome is inherited as an autosomal recessive trait, although a pseudo-dominant Usher syndrome-like phenotype due to a mutation in the mitochondrial MTTS2 gene has been described.12,13 Some reports have also suggested the presence of a possible type IV Usher syndrome with X linked recessive inheritance,14–18 and X linked inheritance could not be excluded in another report of presumed Usher syndrome type I, bronchiectasis, and immotile cilia syndrome.19
We recently identified a family with XLRP associated with hearing loss and recurrent infections of the ear, sinuses, and respiratory tract. Hearing loss and/or recurrent respiratory tract infections have rarely been reported in association with XLRP. In 1992, a family with XLRP and recurrent respiratory tract infections was described.20 This family, which had features indistinguishable from immotile cilia syndrome but no hearing loss, was later shown to harbour a G→T transversion in the 5′ donor splice site of intron 5 of the RPGR gene.21 Another family with XLRP and sensorineural hearing loss, but with no history of recurrent respiratory tract or ear infections, was reported in 1997.22 A 6.4 kbp deletion of the RPGR gene was subsequently identified in this family.5,23 More recently, another group reported a family with XLRP in which hearing loss and recurrent infections coexisted, similar to our family.24 In that case, a 2 bp deletion at position 845–846 of the RPGR gene was identified. We report here an association of the unique phenotype of XLRP, relapsing otitis media, recurrent respiratory tract infections, and hearing loss with a recently reported novel missense mutation of the RPGR gene.7 Furthermore, we demonstrate immunohistochemically the expression of RPGR not only at the retinal level, but also in all extra-ocular tissues targeted by this syndrome in locations consistent with the observed signs and symptoms, providing evidence for a plausible link between RPGR and this newly recognised syndromic phenotype.
Key points
-
We report a family with X linked recessive retinitis pigmentosa (RP) associated with the unique phenotype of relapsing otitis media (ROM), recurrent upper respiratory tract infections (RUTI), and hearing loss, in which a G173R missense mutation in the RPGR gene was identified.
-
In addition to classical RP in males and late onset mild patchy RP with a cone>rod pattern of dysfunction in carriers, audiometry showed mixed hearing loss in affected males with ROM and RUTI, and sensorineural loss in a female carrier who had also suffered from ROM.
-
Using immunohistochemistry, we demonstrated specific RPGR expression in the epithelial lining of human bronchi and sinuses, and in human and monkey cochlea.
-
Our findings provide additional evidence in favour of a broader phenotypic range in association with RPGR mutations than previously suspected and suggest an important role for RPGR in the respiratory tract and cochlea.
SUBJECTS AND METHODS
Subjects
A six generation pedigree of the family is illustrated in fig 1. The proband (VI:1) was a 10 year old white boy seen at our centre for a suspected diagnosis of RP in the context of a previously diagnosed hearing loss. He was referred to us to examine the possibility of Usher syndrome because the family history was positive for this diagnosis in a great great uncle on the maternal side (subject III:4). This history was compounded by a recent diagnosis of “late onset RP” in the maternal grandmother of the proband (subject IV:7). The mother of the proband noted that her youngest son (VI:2) had also been developing hearing loss, seemingly in conjunction with recurrent ear and sinus infections. She also noted that he too might have started to experience the same visual problems as his older brother. There was no known consanguinity in this family, and the mother of the proband was asymptomatic. One aspect of the history that argued against the referral diagnosis of Usher syndrome was that the hearing loss was associated with recurrent ear and sinus infections, which is not a reported feature of Usher syndrome. This initial assessment was most consistent with X linked inheritance and the family was studied accordingly.
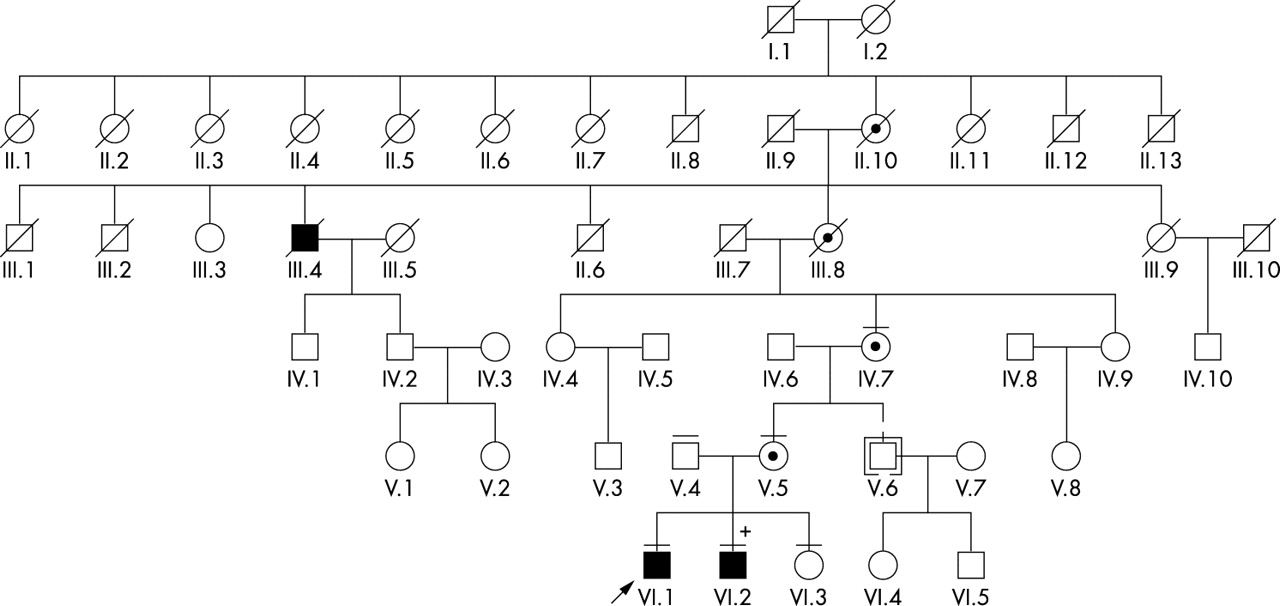
Pedigree of the family. Black symbols identify subjects affected with retinitis pigmentosa (RP) and hearing loss, circles with a black centre identify obligate or ascertained carrier females, and clear symbols identify known or presumed unaffected individuals. + identifies subject VI:2, who was also born with a cardiopulmonary malformation (see text). The horizontal bars above the symbols identify the subjects who were examined clinically and from whom a blood sample was also obtained (except for subjects V:4 and VI:3, from whom only a blood sample was obtained). The arrow identifies the proband in this family. Subject V:6 (symbol in brackets) was adopted into the family.
The proband (VI:1), his younger brother (VI:2), the mother (V:5), and the maternal grandmother (IV:7) were all available for clinical and functional evaluation. After detailed explanation of the procedures and nature of all tests, informed consent was obtained from each patient or their legally authorised representatives to collect a blood sample for DNA extraction and molecular genetic investigations, and to use the findings from the clinical examinations for genotype–phenotype correlation analysis. Blood samples for molecular genetic characterisation were also collected from the father (V:4) and the asymptomatic sister (VI:3) of the proband. All studies were approved by the institutional review boards of the participating institutions, and were in accordance with the Declaration of Helsinki.
Clinical and functional assessment
All patients underwent a detailed history assessment, and a complete clinical and functional ophthalmological investigation, including ERG testing according to international (ISCEV) standards.25 Hearing had been assessed elsewhere by standard pure tone audiometry (PTA) and tympanometry in both VI:1 and VI:2 and, more recently in subjects IV:7 and V:5. These records, along with others concerning previous surgical and diagnostic procedures, were obtained and carefully reviewed. Because of the apparent reliability and completeness of the available systemic data, no further testing was performed at our facilities.
Molecular genetic analyses
Analyses were performed on genomic DNA extracted from whole blood samples collected from the entire nuclear family of the proband and from subject IV:7. DNA was extracted from lymphocytes using standard procedures. Mutation analysis of the RPGR gene was performed as described in detail in Breuer et al.7
Immunohistochemistry
Our objective was to determine if RPGR expression could be demonstrated not only in the retina, but also in other tissues involved in the observed syndrome. Sections from human autopsy specimens of retina, bronchi, and sinuses, and sections of monkey and fetal human cochlea were obtained for immunohistochemical analysis. Two affinity purified polyclonal antibodies raised in rabbit against linear peptides from the RPGR human sequence (kindly provided by Dr P Ferreira, Medical College of Wisconsin, Milwaukee, WI, USA)26 were used in all immunohistochemistry experiments. The RPGR 33606 antibody was raised against a 14 amino acid sequence unique to the RPGR C terminus. The MCW27 antibody was raised against a 22 amino acid terminal sequence unique to exon 15 of the human RPGR open reading frame (ORF15).
All human donor or autopsy tissue specimens were fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) at 4°C for 2–3 weeks, dehydrated in a series of ethanol solutions and xylene baths, and subsequently embedded in paraffin. Monkey cochlear tissue was prepared for paraffin sections by anaesthetising the animal with intraperitoneal injection of urethane (1.5 g/kg), and exsanguinating with a transcardial perfusion of saline with 1% sodium nitrite, followed by fixation with 10% formalin and 1% acetic acid in PBS. The bulla cavity of the animal was quickly exposed and 0.2–0.5 ml of fixative was injected slowly into the scala tympani through the perforated round window. Specimens were kept overnight at 4°C in fixative. All cochlear sections also required decalcification prior to dehydration and embedding. In human fetal tissue, this was achieved via 0.1 mol/l EDTA in 1× PBS at 4°C for approximately 2 weeks.27 For monkey tissues, fixation was followed by 1 week in 120 mmol/l EDTA at pH 7. Serial 8–12 μm sections of the paraffin embedded tissues were cut and mounted on glass slides. Prior to immunostaining, all paraffin sections were deparaffinised, hydrated, and rinsed in deionised water and PBS. Human tissues were obtained following guidelines established by the human research committees and institutional review boards of all participating institutions.
The care and use of animals were in accordance with the NIH Principles of Laboratory Animal Care and approved by the institutional committees on animal care of all participating institutions.
Retina
A human donor eye (65 year old donor; 4.5 hours death to preservation time) was obtained from the Mid-South Eye Bank. Immunostaining of paraffin sections was performed using fluorescent tagged antibodies. All incubation procedures were performed in humid chambers. Non-specific binding sites were blocked with 5% goat serum in PBS for 1 hour at room temperature. The primary anti-RPGR 33606 and anti-RPGR-ORF15 antibodies were diluted to 1:400 and 1:50 respectively, after which time they were applied to the retinal sections for an overnight incubation at 4°C. After washing with PBS, sections were incubated for 1 hour with goat anti-rabbit Cy3 conjugated secondary antibody diluted 1:20 (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Sections were counterstained with 4′, 6′-diamidino-2-phenylindole, dihydrochloride (DAPI; 1 μg/ml final concentration; Molecular Probes, Eugene, OR, USA) to visualise the nuclei. Sections were viewed with a Nikon E800 photomicroscope equipped with the appropriate fluorescent filters. Separate images of the Cy3 and DAPI labels were captured using a SenSys digital camera (Photometrics, Ltd., Tucson, AZ, USA) and superimposed using MetaView software (Universal Imaging Corporation, West Chester, PA, USA). Negative controls involved substitution of the primary antibodies with PBS (data not shown).
Bronchi and sinuses
Sections of a male human principal bronchus (48 year old donor) and of a male human maxillary sinus (16 year old donor) were stained with the RPGR 33606 and the MCW27 antibodies using a protocol identical to that described for the retina.
Cochlea
One adult monkey (unknown age) and one fetal human (20 week old donor) cochlea were used in this study. Immunostaining of paraffin sections was performed with the biotinylated tyramine (BT) enhancement method.28 Sections were blocked with 5% normal horse serum (NHS) in PBS for 30 minutes and then incubated overnight in 1% NHS-PBS at room temperature in a humid chamber with primary anti-RPGR 33606 and anti-RPGR-ORF15 antibodies diluted to 1:400 and 1:50, respectively. Sections were rinsed with PBS and incubated for 1 hour in a 1:1000 dilution of biotinylated goat anti-rabbit IgG (Vector Laboratories, Inc, Burlingame, CA, USA) in 1% NHS-PBS. Sections were rinsed in PBS and incubated with Vectastain ABC reagent (Vector Laboratories). After 1 hour, sections were rinsed in PBS and incubated with BT diluted 1:100 in 0.01% H2O2 for 10 minutes, rinsed in PBS, and incubated with ABC reagent for another 30 minutes. The primary antibody was visualised using 0.05% 3,3′-diaminobenzidine (DAB) in 0.01% H2O2 and 0.1 mol/l phosphate buffer. The negative controls for the immunostaining procedures were done without the application of the affinity purified primary antibody (data not shown).
RESULTS
Clinical and functional ophthalmological findings
Subject VI:1 (proband)
The mother of this 10 year old boy reported a history of visual field limitation, night vision problems and light aversion of recent onset. With moderate myopic correction, his visual acuity could not be improved beyond 20/32−2 with either eye. Fundus findings, which were mainly remarkable for mild perimacular atrophic changes and a punched out appearance of the mid-periphery, are illustrated in fig 2 (panels A and B). ERG testing showed moderate to marked bilateral amplitude reduction and delay of all responses (not shown). In relative terms, there was evidence of symmetric greater rod than cone impairment, consistent with classical RP (rod>cone pattern of dysfunction).

Clinical retinal findings. (A) Posterior pole of the right eye of the proband (subject VI:1), remarkable for a moderate posterior staphyloma, mildly tilted optic discs, and moderate perimacular retinal atrophy (yellowish area) between the optic disc and the fovea. (B) Inferior mid-peripheral retina of the same eye of the proband. Microscopic defects of the retinal pigment epithelium, which were more evident in the inferior hemi-retina, conferred a punched out appearance to the mid-peripheral retinal tissue. This image is also representative of case VI:2. The typical bone spicule-like intraretinal deposits of RP were absent, and only minimal punctate pigmentary deposits could be appreciated (not shown). (C) Posterior pole of the left eye of subject VI:2 (affected male, brother of the proband); a similar but more modest thinning of the perimacular retina between the optic disc and the fovea can be seen. (D) Superonasal mid peripheral retina of the same eye of subject VI:2, illustrating the fine punctate pigmentary deposits that were observed in the retina of these children, more abundant in areas of thinned retinal tissue (discoloured yellowish area). This image is also representative of case VI:1. Microscopic defects of the retinal pigment epithelium, prominent in both the superior and the inferior retinal quadrants, conferred a finely punched out appearance to the mid-peripheral retina in this case also (not shown). (E) Nasal retinal mid periphery of the right eye of subject IV:7 (symptomatic female carrier, grandmother of the affected children). Classical patchy bone spicule-like intraretinal pigmentary deposits can be readily seen. Except for these patches of RP, asymmetrically distributed between the two eyes, the remainder of the eye examination was essentially normal for age. (F) Posterior pole of the right eye of subject V:5 (asymptomatic female carrier, mother of the affected children), which illustrates an essentially normal fundus. This normal appearance was corroborated by normal results to functional testing.
Subject VI:2
This 7 year old boy had behaviour suspicious for visual field limitation of recent onset. His visual acuity could not be corrected to better than 20/32. Fundus examination revealed symmetrical findings that were very similar to those of his brother, although milder in nature (illustrated in fig 2C and D). ERG testing, performed under full anaesthesia, showed the same findings as in subject VI:1 (not shown).
Subject IV:7
This 56 year old woman had a history of myopia and astigmatism since the age of 5 years, with night vision and visual field limitation problems since the age of 50 years. Although this woman could be corrected to approximately 20/20 with each eye, fundus examination disclosed typical bone spicule-like patches of intraretinal pigmentation throughout the mid-peripheral retina (fig 2E), more prominent in the superonasal retina and in the right eye. Consistent with the observed ophthalmoscopic features of patchy asymmetric RP, ERG testing showed mild to moderate asymmetric reduction in amplitude and delayed timing of all responses (not shown). In contrast to the observations in the affected male hemizygotes, there was evidence of greater cone than rod impairment (cone>rod pattern of dysfunction).
Subject V:5
The ocular history of this 38 year old woman was remarkable for hyperopia and astigmatic anisometropia, and non-progressive light aversion of more than 20 years’ duration. Her corrected visual acuity was 20/16 in the right eye and nearly 20/20 in the left anisometropic eye. Her fundus examination was essentially within normal limits (fig 2F), except for a non-specific mottling of the superior mid-periphery. ERG testing was within normal limits in each eye, and no interocular asymmetries were noted (not shown). In summary, the outcome of the ophthalmological investigation in this obligate carrier was within normal limits.
Systemic findings
Subject VI:1 (proband)
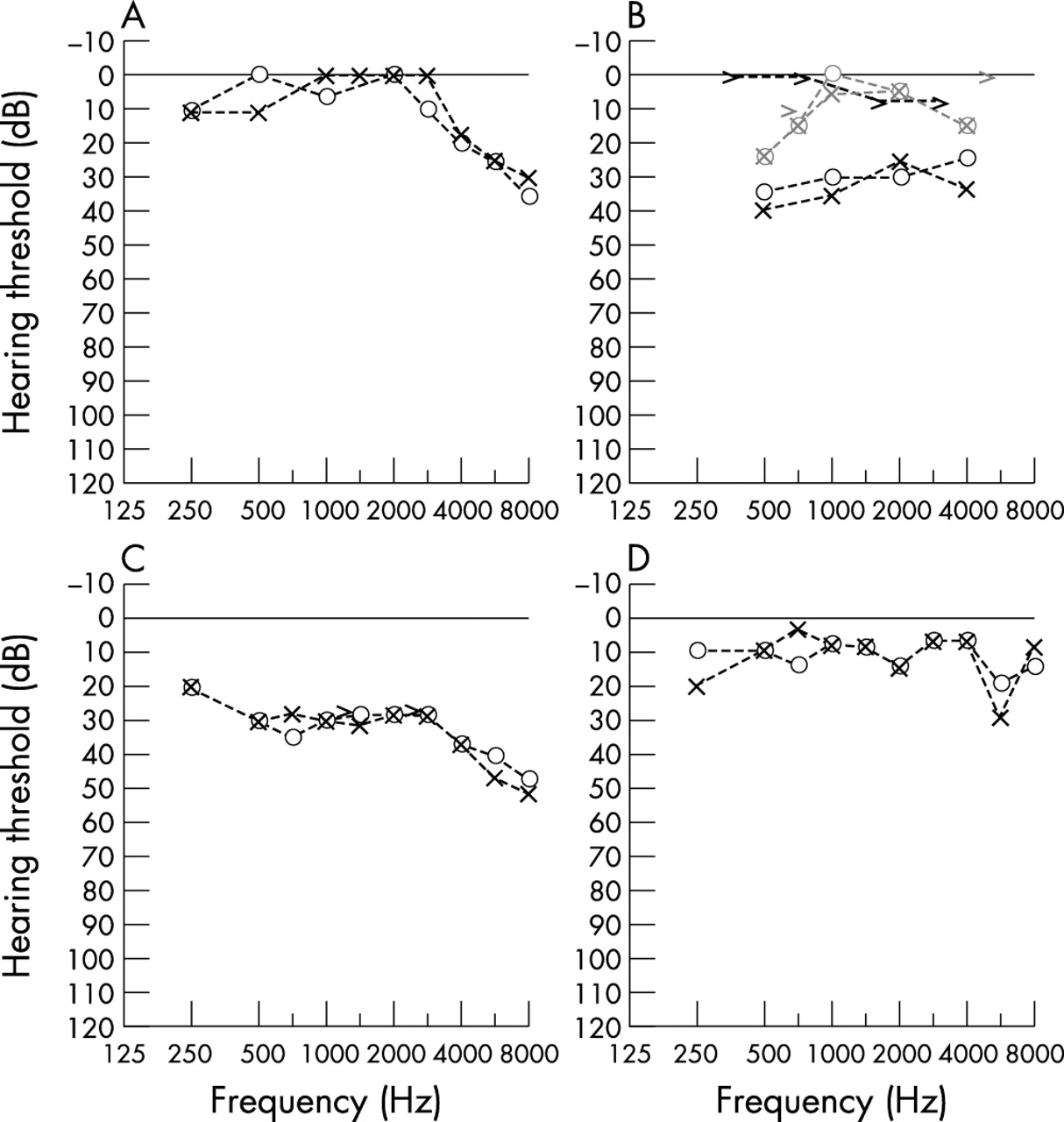
The history of this child was remarkable for numerous episodes of respiratory tract infections, beginning with an episode of bronchiolitis at 10 weeks of age. Recurrent ear infections required repeated myringotomy tube insertion (MTI) bilaterally within the first years of life. Imaging studies also revealed pan-sinusitis at age 3, which was surgically treated with transient benefit. PTA was still reportedly within normal limits in 1999, yet the MTIs could not completely prevent the development of a mild high frequency hearing loss (fig 3A).

Pure tone audiometry (PTA). ○ and ×, aerial conduction, right and left ear, respectively; >, bone conduction, left ear. (A) Subject VI:1 (proband) at 9 years of age, showing a sloping hearing loss at the high frequencies of stimulation, a pattern mainly consistent with a sensorineural origin. (B) Subject VI:2 (affected male, brother of the proband) at 6 years of age before (black symbols) and after myringotomy tube insertion (MTI, grey symbols). The conductive nature of the hearing loss in this subject is revealed by the difference between aerial and bone conduction results. The benefits of the MTI are readily appreciable, although they were reportedly only transitory (no PTA were available subsequent to our visit). Owing to the young age, frequencies greater than 4000 Hz were not tested in this child. (C) Subject IV:7 (symptomatic female carrier), showing a mild sensorineural depression across most frequencies, but more prominent at frequencies greater than 4000 Hz. (D) Subject V:5 (asymptomatic female carrier), which were essentially normal except for a dip at 6000 Hz in the left ear of unclear significance.
Subject VI:2
The array of systemic manifestations in the younger brother of the proband was remarkably similar to that of his sibling. Recurrent ear infections required two MTIs within the first few years of life. In the short run, surgical remedies improved the hearing of this child (fig 3B), but the benefit was reportedly transient. Sweat chloride tests were performed at 4 months and 6 years of age, and in both cases they yielded normal results, thereby providing no evidence for cystic fibrosis. In addition, this child was born with a hypoplastic right lung, right bronchial malacia, stenosis of the left main bronchus, and right pulmonary artery atresia.
Subject IV:7
The systemic complaints of this symptomatic obligate carrier were recurrent ear infections over several decades, progressive hearing loss over the previous 15 years, presently reported as moderate, and excess weight, also of 15 years’ duration. The recurrent infections caused three episodes of tympanic membrane perforation and required mastoidectomy around the age of 30 years. PTA demonstrated (fig 3C) bilateral symmetric mild to severe sensorineural hearing loss with speech discrimination scores of 100%. The tympanograms were normal. Her present height was 153 cm and her weight was 83 kg (body mass index (BMI) 35.5).
Subject V:5
This obligate carrier’s only systemic complaints were a long standing history of anosmia and migraines, and a 10 year history of thyroid problems subsequent to Hashimoto’s thyroiditis. She denied any history of significant noise exposure. Her present height was 162 cm and her weight was 74 kg (BMI 28.2). This BMI elevation reportedly developed and persisted only after the pregnancies and the thyroiditis. She did not complain subjectively of hearing loss and, except for a 6000 Hz dip, PTA revealed an essentially normal result (fig 3D). Speech discrimination scores were 100% binaurally, and tympanograms were normal.
Mutation analysis
A G→C nucleotide substitution at position 576 (G576C) of the RPGR gene (fig 4; nucleotides were numbered according to Meindl et al)4 was identified in the two male hemizygotes (subjects VI:1 and VI:2), and in the heterozygous state in both the obligate carrier asymptomatic mother (V:5) and in the symptomatic carrier maternal grandmother (IV:7). The father of the proband (V:4) and the asymptomatic sister of the proband (VI:3) did not carry the G576C substitution, nor was this found in over 200 normal chromosomes. This substitution is predicted to result in a glycine (non-polar amino acid) to arginine (charged amino acid) change at position 173 (G173R). Gly173 is an invariant amino acid within the RCC1-like homology domain of the RPGR gene product (fig 5). This missense mutation has been reported recently among those detected in a RPGR screening of a large cohort of XLRP families from North America,7 and had not been previously detected.
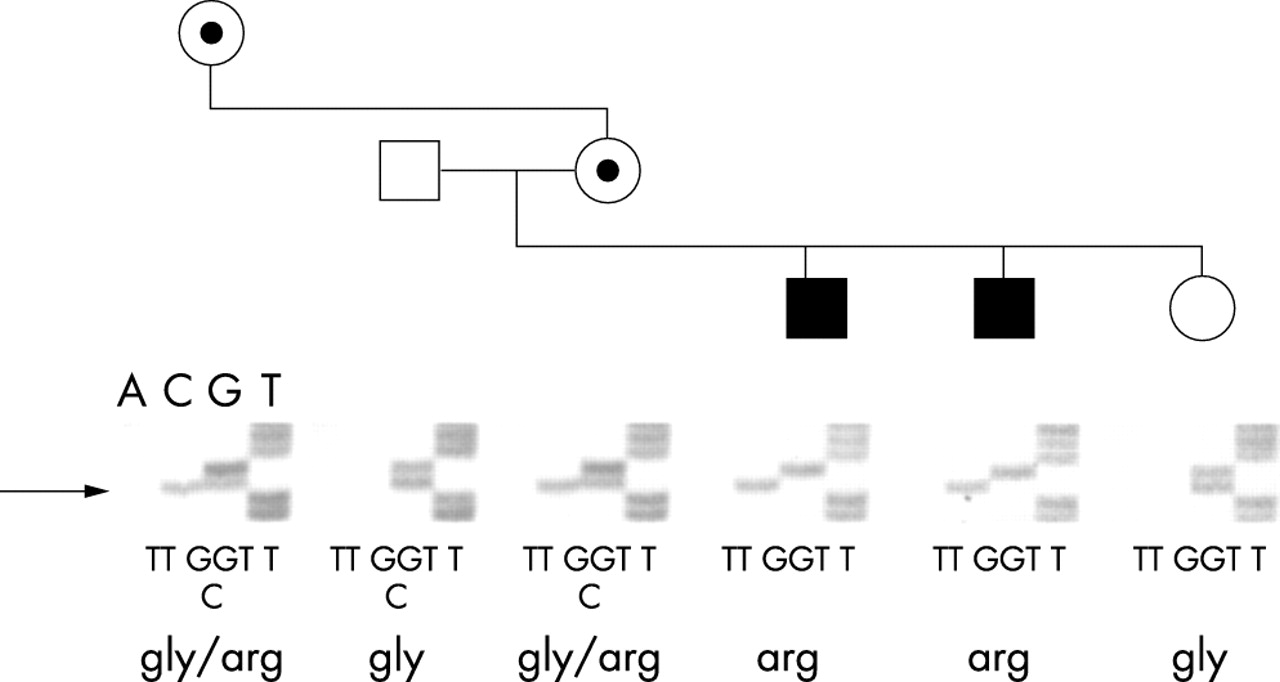
Identification of the RPGR mutation. Results of DNA sequence autoradiographs of the family, showing partial sequence from a forward primer for exon 6, confirm association of sequence alteration with disease. The single base pair change is indicated by the arrow. Coding sequence and translation of the alleles show the amino acid change resulting from the mutation.

Amino acid sequence of human RPGR aligned with that of RCC1 homologues of human, hamster, Drosophila, and two homologues of Saccharomyces. Boxes show regions of conservation while bold amino acids are invariant. The shaded residue in RPGR shows the amino acid altered by the mutation identified in this family.
Immunohistochemical findings
Retina
Consistent with a recent report by Mavlyutov et al,26 immunofluorescence experiments (fig 6A) confirmed expression of RPGR in human retinas not only in the region of the cilium, but also throughout the outer segment of both rod and cone photoreceptors. Results with the 33606 anti-RPGR C-terminus and MCW27 anti-ORF15 antibodies were superimposable, as anticipated from the results of Mavlyutov et al26 and from the fact that both peptidic sequences are known to be expressed in the retina.

Immunofluorescence images of sections from human retina (A), bronchi (B) and sinuses (C). The red areas indicate immunopositive staining for RPGR, whereas the green areas indicate the location of nuclei (DAPI staining). Results obtained with the antibodies generated against the C-terminus of RPGR (33606) and the ORF15 (MCW27) were identical. For brevity, only those results obtained using the anti-ORF15 antibody are shown in this figure. (A) In the retina, ORF15 immunopositive labelling is limited to the photoreceptor outer segment area where it is expressed at very high levels. (B) An equally high level of ORF15 immunopositive labelling is present in the bronchi. In this tissue, the labelling is restricted to the epithelial cells that line the lumen. (C) The anti-ORF15 antibody similarly recognised an antigen localised exclusively to the epithelial cells lining the sinus cavities. RPE, retinal pigment epithelium; OS, photoreceptor outer segments; IS, photoreceptor inner segments; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Asterisks (*) in Panels B and C identify the lumen of the bronchus and sinus, respectively. Scale bar: 100 µm.
Bronchi and sinuses
Similar to retinal experiments, anti-RPGR immunofluorescent staining of human bronchial and sinus sections revealed a very specific and discrete localisation of reactivity to the epithelial lining of both tissues (fig 6B–C). However, contrary to the assumption that ORF15 is a retina specific exon, identical immunostaining patterns were observed with both antibodies also in these tissue samples.
Cochlea
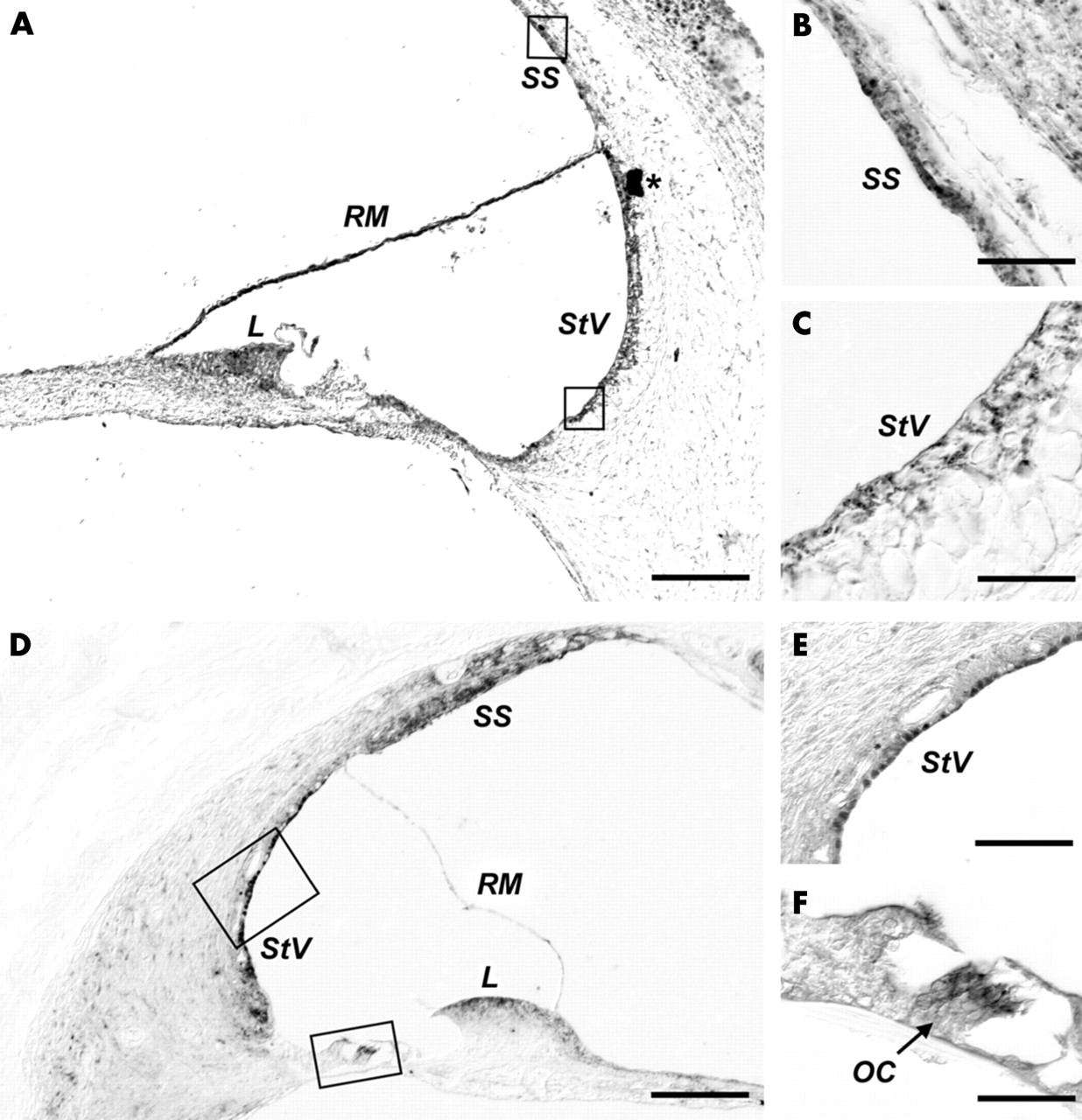
Immunostaining with both the 33606 anti-RPGR C-terminus and MCW27 anti-ORF15 antibodies showed in the human fetal cochlea strong, consistent localisation to the stria vascularis, to the suprastrial cells, and to the apical portion of the spiral limbus (fig 7A–E). In the human fetal tissue sections, the staining pattern of the 33606 anti-RPGR and MCW27 anti-ORF15 antibodies were virtually identical (not shown). Immunostaining experiments were replicated in monkey with the 33606 anti-RPGR C-terminus antibody. Some differences between human and monkey were observed. For example, the patchy immunoreactivity that was seen in the Reissner’s membrane in the fetal sample (fig 7A, detail not shown) was not observed in monkey (fig 7D). Conversely, in the monkey section, in addition to persistent staining at the level of the stria vascularis, suprastrial cells, and limbus, immunoreactivity was also seen in the organ of Corti (fig 7F); however, this could not be ascertained in the fetal human section owing to lack of tissue preservation in that region and perhaps also the difference in fixation solution used. Therefore, it could not be determined with certainty whether this finding could also be generalised to human tissues.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cochlear immunostaining patterns. Positive immunostaining is shown in all panels as the dark DAB reaction product. (A) Immunohistochemistry with the anti-ORF15 (MCW27) antibody in a cross-section of a fetal cochlear turn fixed in formalin. Immunoreactivity is noticeable at the level of the stria vascularis (StV), the suprastrial (SS) cells, the apical portion of the spiral limbus (L), and in a patchy fashion along the Reissner’s membrane (RM). (B–C) Close-up view of the boxed regions in (A). (B) Detail of the immunoreactivity to anti-ORF15 antibody in the SS cells. Identical results were obtained with the anti-RPGR 33606 antibody (not shown). (C) Detail of the cells of the StV stained by the anti-ORF15 antibody. (D) Immunohistochemistry using the anti-RPGR 33606 antibody in a cross-section of one cochlear turn of formalin acetic fixed monkey tissue. (E, F) Close-up view of the boxed regions in (D), showing RPGR immunolocalisation to the StV and to the organ of Corti (OC), respectively. Scale bars: (A, D) 100 µm; (B, C, F) 12.5 µm; (E) 25 µm.
DISCUSSION
The array of clinical manifestations observed in this family exceeded that of classical XLRP and, because of the associated hearing loss, led to the initial incorrect referral diagnosis of suspected Usher syndrome. However, results of our investigations unveiled a more complex phenotype. The mild degree and non-congenital nature of hearing loss in the family was revealed by the absence of speech abnormalities in the affected children, which is typically noticeable in all patients with Usher syndrome type II. While the hearing loss may be progressive in this form of XLRP, the presence of a conductive component that was improved by MTI, and the association with the recurrent infections distinguishes it from that of Usher syndrome type III. Despite these differences, our findings emphasise the need for inclusion of XLRP among the differential diagnoses of presumed Usher syndrome, and for the hearing loss history to be carefully characterised.
Despite the relative subtlety of the ophthalmoscopic changes, the form of retinal degeneration affecting the two male children in this family was typical for the classic rod>cone dystrophy pattern of RP. In contrast, subject IV:7, a female carrier, had ERG changes consistent with greater cone than rod dysfunction. Cone predominant phenotypes have been reported previously both in female heterozygotes29,30 and in male hemizygotes31–33 carrying RPGR mutations, thereby suggesting an important role for RPGR in both rods and cones. This hypothesis is consistent with the immunohistochemical evidence of expression of RPGR in both types of photoreceptors.26,34
Our clinical findings and those of Zito et al35 extend the phenotypic spectrum of X linked disease associated with RPGR mutations, already inclusive of RP,4,5 cone-rod dystrophy,31,32 macular degeneration,33 and even a quasi-dominant X linked RP in families with ORF15 mutations,36, to the syndromic range. Our immunohistochemical data (figs 6 and 7) provide for the first time evidence of specific RPGR expression in the human epithelial lining of the respiratory epithelia and at the cochlear level, locations consistent with the array of symptoms presented by this family. Our findings in the respiratory tract are also consistent with the low levels of RPGR expression previously reported in lung tissue by several investigators.4–5,37–39 While these findings do not explain why, to date, only our family and that of Zito et al35 have been diagnosed with this broader phenotype, they provide a solid rationale for considering the syndromic phenotype herein reported as a the likely result of impaired RPGR function in affected tissues. Furthermore, our data and those of Zito et al,35 combined with previous published evidence of an association of RPGR dependent XLRP with hearing loss,22 suggest that the previously hypothesised existence of an Usher syndrome type IV may be best explained by XLRP associated with hearing loss and RPGR mutations.
It is unclear whether the congenital pulmonary abnormalities that affected case VI:2 and the anosmia that affected case V:5 have any relationship to the observed syndromic phenotype. However, no malformation of the pulmonary system was present in any other member of this family. Smell function was tested with the University of Pennsylvania Smell Identification Test for subject V:5, and the results were indeed consistent with complete anosmia of the congenital type, whereas testing of case IV:7 was entirely normal (not shown). The affected males were too young to be tested reliably. Therefore, while a possible link between these findings and the observed syndrome cannot be entirely excluded, they presently appear to be coincidental.
The observed hearing loss had a conductive component at least at some point in the course of the disease and was amenable to surgical improvement (fig. 3B). However, MTIs could not prevent the development of sloping sensorineural hearing loss. This pattern was observed both in subjects VI:1 and IV:7 (fig. 3). It is difficult to rule out with certainty whether the latter was contributed by early onset presbyacusis, but the onset around the age of 40 years without a history of noise exposure in the context of this syndrome make presbyacusis an unlikely explanation. In addition, it is difficult to establish whether the relapsing ear infections contributed to the hearing loss in this subject. Despite these uncertainties, there are previous reports of non-infectious, pure sensorineural hearing loss in XLRP families with RPGR mutations.22 This indicates that a history of infections is not a prerequisite to suspect XLRP with hearing loss in the differential diagnosis of Usher syndrome. Indeed, although still preliminary, our immunohistochemical findings show that RPGR is expressed in the cochlea. Specifically, consistent expression in both monkey and fetal human tissue samples was observed in stria vascularis, in the suprastrial cells, and in the spiral limbus. These structures share high metabolic activity, appear to be involved in active transport functions, and are presumed to participate in endocochlear potential maintenance and endolymphatic K+ recycling.40–44 If RPGR expression were indeed also present in the human organ of Corti as suggested by the monkey data, this too would represent another possible pathway through which sensorineural hearing loss could occur. Therefore, the observed immunolocalisation patterns appear entirely consistent with the symptoms experienced by our family and in others with known RPGR mutations,22,35 and suggest that defective RPGR function may be implicated in determining the observed hearing loss.
Although RPGR is ubiquitously expressed and produces numerous alternate splicing isoforms that are differentially expressed across tissues,4,37,38 the function of RPGR in extra-ocular tissues is essentially unknown because RPGR until now has been believed to be associated only with a retinal phenotype. Even in the retina, the localisation and function of RPGR remains incompletely understood. In mice, elegant studies have localised RPGR exclusively to the region of the photoreceptor connecting cilium,45,46 where it appears to be anchored via its interaction with RPGRIP1.45,47 This suggested that RPGR might be specifically involved in the trafficking of rhodopsin and/or other phototransduction proteins along the cilium from the inner to the outer segment of the photoreceptor. Recurrent and chronic otitis media with associated conductive hearing loss are common sequelae of middle ear cleft mucosal abnormalities such as ciliary dysfunction. Therefore, it is possible that the RP and recurrent infections observed in this family and those reported by Van Dorp et al20 and Zito et al35 might be due to impaired ciliary function in both the retina and the respiratory epithelium. This hypothesis may well explain the ciliary abnormalities of the nasal mucosa and spermal axonemes observed particularly in patients with XLRP.48–50
However, more recent studies with a different panel of antibodies have shown important differences in the patterns of RPGR expression in the human retina, where RPGR also appears to be expressed throughout the outer segments of both rod and cone photoreceptors.26 With these same antibodies, we too have confirmed this broader pattern of expression in human retinal photoreceptors. Therefore, RPGR functions also may be broader than ciliary trafficking alone. Likewise, ORF15 had been suggested to be a relatively “retina specific” sequence;39 however, by using the MCW27 anti-ORF15 antibodies, we showed highly specific expression of this sequence at least in the epithelial lining of the respiratory tract and in non-ciliated cochlear tissues, which suggests a wider expression of ORF15 and a broader functional role of RPGR than previously recognised. We are unaware of any reported cochlear abnormality or respiratory tract problem either in the RPGR knockout mouse34 or in the Siberian husky, which harbours naturally occurring RPGR mutations.51 However, further preliminary immunohistochemical studies on mouse cochleas (not shown, unpublished data) with polyclonal anti-mouse antibodies raised towards the C-terminal region of RPGR34 showed staining patterns that were only partially consistent with our human and monkey data. Therefore, interspecies differences may, at least in part, preclude a full appreciation of the physiological roles of RPGR. This issue, however, warrants further investigation.
The glycine at codon 173 mutated in this family is a highly conserved residue of the RCC1-like domain of RPGR. RCC1 is a guanine nucleotide exchange factor that regulates the GTPase Ran, a ubiquitously expressed protein with several proposed functions, including GTPase coupled membrane transport and trafficking.4 The change from glycine to the charged amino acid arginine (G173R) is likely to disrupt the normal function of this domain. We do not have pathological evidence to explain why recurrent ear and respiratory infection, hearing loss, or both have been reported only in association with a limited number of families with mutations in RPGR. Based on the aforementioned evidence, one possible explanation may be that certain mutations affect regions of the RPGR gene product that are important not only to the photoreceptors but also to other tissues, such as the respiratory mucosa and the cochlea, while others may not. It is possible that the G173R missense mutation detected in this family caused a gain of function effect, thereby leading to unique anomalous effects in other tissues in the body in which RPGR is also expressed. Alternatively, disruption of the RCC1-like domain caused by the G173R mutation may be as severe as that caused by truncating changes such as those seen in other families,5,22–23,35 and lead to severe loss of function outside of the retina as well. Lastly, it cannot be excluded that the variability of the phenotypes across families may also depend on co-inheritance of polymorphic variants of other genes, and specifically of RPGR interacting proteins (RPGRIPs), which await identification. Whatever the explanation, distinct RPGR mutations do appear to be prone to yielding very different phenotypes, even within the same tissue, as in the case of the macular degeneration phenotype recently reported by Ayyagari et al,33 which is in striking contrast to the retina wide phenotypes thus far reported by all other investigators. Further clinical, laboratory, and genetic epidemiological studies will hopefully provide an explanation for these aspects in the not too distant future.
In conclusion, this report and that of Zito et al35 provide additional evidence in favour of a broader phenotypic range in association with RPGR mutations than previously recognised, and suggest an important role for RPGR in extra-ocular tissues, in particular the respiratory mucosa and the cochlea, a role that was not previously appreciated. Specifically, our findings, those of Zito et al,35 and those of van Dorp et al20 suggest an important role of RPGR in ciliary physiology, and perhaps more generally in processes requiring motility and/or active transport. However, the understanding of the function(s) of RPGR and its RCC1-like homology domain remains largely incomplete. Further studies will be necessary to characterise in full the spectrum of manifestations that can be associated with RPGR mutations and understand the function(s) of RPGR across tissues. Accordingly, until further studies across species are conducted, the generalisation of any understanding of the role of this protein from animals to humans may warrant caution.
Acknowledgments
We gratefully acknowledge grant support to this investigation from the following sources: Le Bonheur Children’s Medical Center Research Grant Program, Memphis, TN, USA (A Iannaccone); University of Tennessee Medical Group Research Grant Program, Memphis, TN, USA (A Iannaccone); National Institute on Deafness and Other Communication Disorders (subcontract to A Iannaccone and M M Jablonski from Boys Town National Research Hospital, Omaha, NE, USA, to Program Project Grant P01 DC01813-07; and grants DC03402 to C C Morton and T32DC00038 to S F Kuo); UTHSC Neuroscience Postdoctoral Fellowship Award (X F Wang); National Eye Institute (UTHSC Ophthalmology Core Grant EY013080 and Grants EY07961 and EY07003 to A Swaroop); Foundation Fighting Blindness, Hunt Valley, MD, USA (A Swaroop); and Research to Prevent Blindness, Inc., New York, NY, USA (Career Development Award to A Iannaccone, William and Mary Greve Special Scholar Award to M M Jablonski, Lew Wasserman Merit Award to A Swaroop, and unrestricted grants to the UTHSC and UM Departments of Ophthalmology).
The anti-RPGR 33606 and MCW27 antibodies used in this study were a generous gift of Dr P A Ferreira, Medical College of Wisconsin, Milwaukee, WI, USA. The anti-mouse RPGR antibodies were a generous gift of Dr T Li, Berman-Gund Laboratory, Massachusetts Eye and Ear Infirmary, Boston, MA, USA. The monkey cochlea section was a generous gift from Dr J C Adams, Department of Otolaryngology, Massachusetts Eye and Ear Infirmary, Boston, MA, USA. The human donor eyes used in this study were kindly contributed by the Mid-South Eye Bank, Memphis, TN, USA.
We would also like to express our deepest gratitude to the patients, who participated enthusiastically in this project.
REFERENCES
Footnotes
-
1 Present addresses: Università Campus Biomedico School of Medicine, Rome, Italy;
-
2 Department of Biochemistry and Molecular Biology, University of British Columbia, Vancouver, Canada