Article Text

Statistics from Altmetric.com
Definition of terms
Pulmonary hypertension is defined as a mean pulmonary artery pressure > 25 mm Hg at rest or 30 mm Hg with exercise.
The classification of pulmonary hypertension is linked to the anatomy or the aetiology. These diagnostic categories are for the convenience of deciding on treatment.
Primary pulmonary hypertension refers to pulmonary hypertension for which no cause can be identified. The term remains unchanged from the previous system of classification. Primary pulmonary hypertension is a form of pulmonary arterial hypertension which also includes collagen vascular disease*, congenital systemic to pulmonary shunts*, portal hypertension*, HIV infection*, exposure to various drugs or toxins*, and persistent pulmonary hypertension of the newborn*. Other types of pulmonary hypertension include pulmonary venous hypertension*, pulmonary hypertension associated with disorders of the respiratory system and/or hypoxaemia*, pulmonary hypertension caused by chronic thrombotic and/or embolic disease*, and miscellaneous causes*.
*These hitherto have been called secondary pulmonary hypertension, a term without value for diagnosis and decisions on treatment.
Synopsis
PRESENTATION AND INVESTIGATION
- 1.
- The symptoms of pulmonary hypertension are relatively non-specific. Breathlessness is the most common.
- 2.
- Where the diagnosis of pulmonary hypertension is suspected a transthoracic echocardiogram should be performed to screen for pulmonary hypertension.
- 3.
- Referral of patients to a designated specialist centre should normally be made after an ECG, chest x ray, simple spirometry, and demonstration of pulmonary hypertension by echocardiography, but before cardiac catheterisation. Referral should not be delayed owing to the risk of early death from this condition.
- 4.
- The diagnosis should be confirmed at right heart catheterisation.
- 5.
- The aetiology of pulmonary hypertension should be sought in order to determine optimal treatment.
- 6.
- Acute vasodilator testing should be undertaken at the time of cardiac catheterisation. The response to acute vasodilator testing accurately identifies patients who may respond to long term oral vasodilator treatment.
TREATMENT
- 7.
- All patients with primary or thromboembolic pulmonary hypertension should be treated with warfarin. Warfarin should be seriously considered in other types of pulmonary arterial hypertension where there are no contraindications.
- 8.
- Vasodilator therapy with oral calcium antagonists may improve symptoms, haemodynamics, and survival in selected patients with pulmonary arterial hypertension who respond to an acute vasodilator test.
- 9.
- Patients with severe pulmonary hypertension which is primary, familial or caused by anorectic agents, connective tissue diseases, shunts associated with congenital heart disease, portal hypertension, sarcoidosis, HIV or chronic thromboembolic disease (either inoperable or as a bridge to pulmonary thromboendarterectomy) should be considered for long term intravenous infusion of prostaglandins.
- 10.
- Patients with chronic proximal pulmonary thromboembolic disease should be considered for pulmonary thromboendarterectomy.
- 11.
- Lung or heart lung transplantation should be considered in selected patients with pulmonary hypertension with disease which is severely symptomatic and progressive despite optimal medical and/or surgical treatment.
- 12.
- Controlled oxygen therapy may be indicated for those patients with sustained nocturnal hypoxaemia where arterial oxygen saturations are below an average of 90% on air and patients with chronic pulmonary disease associated with hypoxia and pulmonary hypertension.
- 13.
- Atrial septostomy may be considered in severe pulmonary hypertension refractory to prostaglandin therapy particularly if it is associated with recurrent syncope.
- 14.
- Women of child bearing age with pulmonary hypertension require contraceptive advice.
- 15.
- Patients should receive a one-off pneumococcal immunisation and annual immunisation against influenza.
- 16.
- Patients with pulmonary hypertension require life long monitoring in a specialist centre with the instigation of appropriate therapies as the disease evolves. Submaximal exercise testing is a useful objective assessment since exercise capacity and severity of pulmonary hypertension are correlated. Patients with pulmonary hypertension caused by hypoxia, chronic heart failure, and congenital heart disease will be followed up by other physicians and do not normally require monitoring in a specialist pulmonary hypertension centre.
Primary pulmonary hypertension (PPH) is a rapidly progressive disease with an incidence of 1–2 per million per annum.1 2 It causes disabling symptoms and untreated leads to early death.3 Other causes of pulmonary arterial hypertension may account for a further 1–2 cases per million per annum. Until 1981 when heart–lung transplantation was introduced there was no treatment. Challenged by the limited numbers of suitable donors, medical treatments have been sought, the most successful of which have now postponed the need for transplantation.
The purpose of this paper is to provide guidance for best practice in the management of adults and children following the practice in those centres which routinely investigate these patients. We present a description of the methods for achieving an accurate diagnosis and determining which patients should benefit from medical and surgical treatments. The recommendations were originally devised for patients with PPH, but they also can be used when considering treatment modalities for the many other forms of pulmonary hypertension. Except where specifically stated, the recommendations for treatment and follow up apply to PPH. We recognise that evidence is lacking in several areas, that the treatment of pulmonary hypertension is evolving rapidly and that there is much still to be learned. Advances in the management of this disease indicate that these recommendations will need to be updated in a few years time.
Patients should be managed by designated specialist centres with experience in investigation and provision of complex forms of treatment. Such centres are expected to be managing at least 40 patients. In view of the poor prognosis in untreated patients referral should not be delayed.
Nomenclature
The pathological classification of pulmonary vascular disease does not correlate with clinical or haemodynamic features of pulmonary hypertension and it provides no useful clue to the aetiology. In 1998 the World Health Organization sponsored a world symposium on pulmonary hypertension. Among a number of important outcomes from this meeting was an agreed new classification of pulmonary hypertension to encompass fresh knowledge on the treatment and the pathogenesis of pulmonary vascular disorders. This has allowed a rational approach to drawing up guidelines for treatment.
The basis of the new classification is the notion of common pathophysiological processes involving the pulmonary blood vessels in all forms of the disorder. The constituent cells of the vessel walls appear to undergo changes in phenotype which in turn alter their structure and function. Certain forms of pulmonary hypertension appear to respond specifically to chronic prostaglandin therapy, while in others the disease state is worsened by these drugs.
This has led to a descriptive classification of the disorder (table1).4 Each class is also defined according to the presence or absence of additional and concurrent disease. Where the pulmonary arteries are the site of the pathology this is classified as pulmonary arterial hypertension. This includes patients with PPH, liver disease, HIV infection, and anorectic treatment. Pulmonary venous hypertension as the name implies is the form of disease which afflicts the pulmonary veins. The most common form of this is seen with left ventricular failure. Hypoxic lung disease causing pulmonary hypertension is another category, as is chronic thromboembolic disease. A fifth miscellaneous group includes other rare causes.
Classification of pulmonary hypertension according to the World Health Organization 1998
Each patient is further classified according to the degree of functional disturbance, based upon exercise performance, graded I–IV in a manner analogous to the New York Heart Association grade (table2).
Functional classification of pulmonary hypertension modified after the New York Heart Association functional classification according to the World Health Organization 1998
Objectives and priorities for investigation
The aim of investigation is to confirm the diagnosis of pulmonary hypertension, determine the type of disease according to the new classification, assess the suitability of possible treatments, and estimate prognosis.
CLINICAL PRESENTATION
The symptoms of pulmonary hypertension are relatively non-specific although they are clearly cardiorespiratory. Breathlessness is most common, followed by chest pain and syncope. Frequently there is a delay of up to three years between the first symptom and the diagnosis,3 and this interval has remained the same over the last 10 years.
Perhaps the best advice is to exclude common respiratory disease as the cause of dyspnoea and simple spirometry is a useful functional screening test to identify primary lung disease. A chestx ray and ECG should be performed since these tests are abnormal in 85% of patients presenting with symptoms caused by established pulmonary hypertension (figs 1 and2).
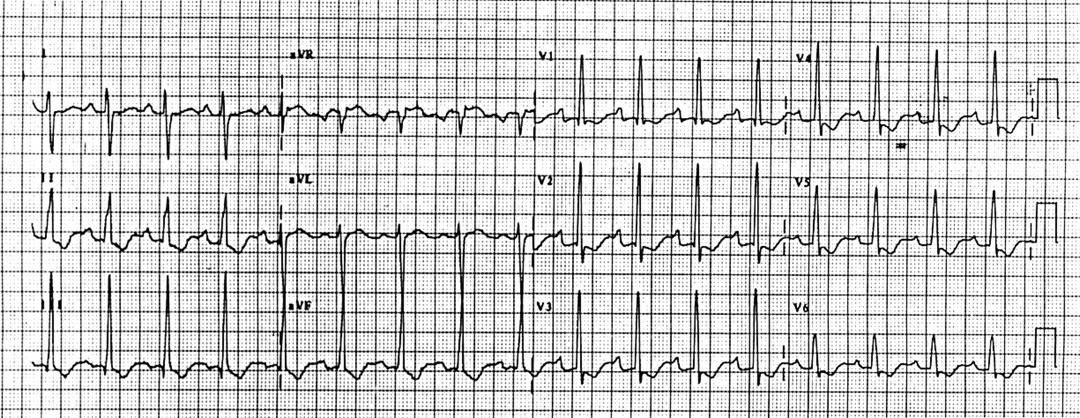
Typical ECG of chronic pulmonary hypertension showing right ventricular hypertrophy.

Chest radiograph of a patient with primary pulmonary hypertension. Note the enlarged proximal pulmonary arteries.
Symptom severity is assessed by exercise testing using a submaximal test such as the six minute walk test5 or incremental shuttle test.6
DIAGNOSIS AND REFERRAL
Where the diagnosis of pulmonary hypertension is suspected a transthoracic echocardiogram should be performed to screen for pulmonary hypertension by estimation of the systolic pulmonary artery pressure from the tricuspid regurgitant jet and the jugular venous pressure.7 Other indirect echocardiographic indications of pulmonary hypertension may also be detected and are summarised in table3.
Some echocardiographic measurements used in pulmonary hypertension
The diagnosis should be confirmed at right heart catheterisation when measurement of right heart pressures, mixed venous oxygen saturation, cardiac output, and response to vasodilators (see “Acute vasoreactivity study”) can be made. In some patients left heart catheterisation will also be indicated.
Referral of patients to a designated specialist centre should normally be made after an ECG, chest x ray, simple spirometry, and demonstration of pulmonary hypertension by echocardiography, but before cardiac catheterisation. Referral should not be delayed owing to the risks of early death from this condition.
AETIOLOGY
The aetiology should be sought in order to determine optimal treatment. An important cause of pulmonary hypertension has been appetite suppressants.8
GENETICS
Most cases of primary pulmonary hypertension are sporadic but 6% are familial.1 The disease is inherited as an autosomal dominant with incomplete penetrance9 10 and shows anticipation with presentation at a younger age in successive generations.11 The gene has been mapped to chromosome 2q 3312 and recently identified as a mutation of the BMPR2 gene, which encodes the type II bone morphogenetic protein receptor (BMPR-II). It exhibits mutations which predict a truncated or dysfunctional protein.13 14 Mutations of the gene encoding BMPR-II are also seen in at least 26% of sporadic cases of PPH.15
There are at least 20 known families in the UK with this condition. The low penetrance of the gene results in a low yield obtained by screening asymptomatic family members.
INVESTIGATIONS
Table 4 shows a list of investigations required in all new patients presenting to a specialist centre with this condition.
Investigations recommended in the assessment of pulmonary hypertension
Lung biopsy is not normally required since the procedure carries significant mortality and except in rare instances does not alter management. In patients with a ventilation perfusion scan suggestive of pulmonary embolism or an equivocal scan with a suspicious history, pulmonary angiography is now supplemented by helical computed tomographic (CT) angiography for the diagnosis and localisation of chronic thromboembolic disease.16 Specialist radiological experience is required to obtain angiograms of sufficient quality if surgical thromboendarterectomy is being considered. Magnetic resonance pulmonary angiography may also provide useful information although its role is as yet unproven.
ACUTE VASOREACTIVITY STUDY
The initial response to acute vasodilator testing accurately identifies patients who may respond to long term oral vasodilator treatment (fig 3).17 It may be undertaken as part of cardiac catheterisation since invasive monitoring is required. The haemodynamic response to intravenous adenosine infusion,18inhaled nitric oxide,19 intravenous epoprostenol or nebulised epoprostenol20 is recorded. Although any of these vasodilators are acceptable, it is expected that the use of inhaled nitric oxide will become standard practice in all centres in the future.

Management of pulmonary arterial hypertension. CI, cardiac index; SvO2, pulmonary arterial oxygen saturation; NYHA, New York Heart Association.
A positive response is present when there is a > 20% reduction in mean pulmonary artery pressure or pulmonary vascular resistance,21 providing there is no fall in cardiac output. This is achieved in about a quarter of patients at presentation. A negative response is recorded when there is no significant improvement or deterioration in pulmonary haemodynamics.
PROGNOSIS
Survival of untreated PPH patients in the National Institutes of Health (NIH) Registry was a median of 2.8 years after diagnosis,3 although with new treatments this has improved (table 5). The individual patient's prognosis can be accurately predicted from the haemodynamic variables which reflect right heart failure (notably pulmonary arterial oxygen saturation, cardiac output, and mean right atrial pressure),3 22-25 severity of symptoms,3 17 exercise capacity measured by six minute walk test,5 26 and response to vasodilators (table6).27 It should be appreciated that although right atrial pressure is a correlate of survival, it may be significantly modified by the use of diuretics. Haemodynamic indices should not be interpreted in isolation.
Adverse prognostic indicators in primary pulmonary hypertension
Objectives and priorities for treatment
MEDICAL THERAPY
Anticoagulation
Pulmonary hypertension is associated with pulmonary arterial thrombosis and a hypercoaguable state associated with a fibrinolytic defect and haemostatic disturbance.28 29 Anticoagulation may reduce thrombosis and slow progression of some forms of the disease. Warfarin therapy has been shown to almost double three year survival in primary pulmonary hypertension.22 30 There are no published data on the use of warfarin in other forms of pulmonary hypertension. Caution regarding anticoagulant use in connective tissue disease is outlined under “Disease specific recommendations”.
Recommendation: All patients with primary or thromboembolic pulmonary hypertension should be treated life-long with warfarin to achieve an international normalised ration (INR) of 2–3. Warfarin should be seriously considered in other types of pulmonary arterial hypertension where there is no contraindication, such as gastrointestinal haemorrhage, significant haemoptysis or liver disease with coagulation abnormalities.
Oxygen therapy
Hypoxaemia causes pulmonary vasoconstriction and may worsen pulmonary hypertension. Continuous oxygen causes a modest reversal of pulmonary hypertension in patients with chronic lung disease31 in whom its use is recommended,32and there is evidence of symptomatic improvement in children in pulmonary arterial hypertension.33 Many patients with pulmonary arterial hypertension show significant falls in arterial oxygen saturation at night. This can be corrected with long term oxygen therapy (2 l/min oxygen from an oxygen concentrator).
Recommendation: Patients with pulmonary hypertension should undergo nocturnal oxygen saturation monitoring. Controlled oxygen therapy may be indicated for those patients with sustained nocturnal hypoxaemia where arterial oxygen saturations are below an average of 90% on air and are corrected on controlled supplemental oxygen.
Supportive medical therapy
Right heart failure gives rise to fluid retention which is improved by diuretics. Digoxin has been shown to improve cardiac output acutely in primary pulmonary hypertension.34 Its efficacy when administered chronically in this condition is unknown although it improves symptoms and reduces hospital admissions without increased mortality in patients with chronic heart failure.35
Recommendation: Diuretics are indicated to control fluid retention. Digoxin may be considered in patients who remain symptomatic on medical therapy.
VASODILATOR THERAPY
The rationale for vasodilators is based on the importance of vasoconstriction in the pathogenesis of pulmonary hypertension.36 Vasodilator therapy with calcium antagonists may improve symptoms, haemodynamics, and survival in pulmonary arterial hypertension. Since this treatment can result in rapid clinical deterioration it should only be used in patients with a positive acute vasodilator study and a cardiac index > 2.1 l/min/m2.
The systemic use of vasodilators in patients with pulmonary venous hypertension may cause pulmonary oedema37 38 by increasing pulmonary blood flow in the presence of downstream obstruction. Vasodilators are also contraindicated in patients with hypoxic pulmonary hypertension caused by chronic lung disease or interstitial lung disease who are prone to worsening hypoxaemia with vasodilator therapy caused by ventilation perfusion mismatch.
Calcium antagonists
Calcium antagonists cause pulmonary and systemic vasodilatation. They are effective in the presence of vasoconstriction but not in its absence. Thus patients who benefit are those with a positive acute vasodilator response. Their acute vasodilator response tends to be maintained long term.30 39 Long term use of high dose diltiazem and nifedipine reduces pulmonary artery pressure40 and mortality with sustained improvement of symptoms.30
Calcium antagonists cause systemic hypotension and their negative inotropic effect may be adverse when right ventricular function is already damaged.41 42 Survival is only improved by calcium antagonists in patients with a right atrial pressure < 10 mm Hg.43 In patients with advanced left ventricular dysfunction diltiazem and nifedipine may worsen heart failure and increase mortality.44-46 Amlodipine is an alternative dihydropyridine which does not increase morbidity or mortality in chronic heart failure.47 It causes pulmonary vasodilatation when administered acutely in pulmonary hypertension48 although its long term effects have not been investigated.
Recommendation: Calcium antagonists should not be started before performance of an acute vasodilator study. For patients with a cardiac index > 2.1 l/min/m2, and/or mixed venous oxygen saturation > 63%, and/or right atrial pressure < 10 mm Hg, and with a positive response to acute vasodilator challenge, a calcium antagonist should be initiated. Diltiazem or nifedipine are appropriate unless right ventricular function is impaired when amlodipine should be considered. Calcium antagonists should be commenced in hospital where they should be uptitrated according to symptoms, blood pressure, oxygen saturation, and exercise tolerance.
For patients with a cardiac index > 2.1 l/min/m2 and/or pulmonary arterial oxygen saturation > 63% and/or right atrial pressure < 10 mm Hg, and who do not manifest responsiveness to acute vasodilator challenge, oral calcium channel blocker therapy is unlikely to be beneficial and may have adverse effects.49
In the presence of cardiac index ⩽ 2.1 l/min/m2 and/or pulmonary arterial oxygen saturation ⩽ 63% and/or right atrial pressure ⩾ 10 mm Hg, calcium antagonists should be avoided.
Long term prostaglandin therapy
Prostaglandins are potent endogenous vasodilators which inhibit platelet aggregation50 and have antiproliferative51 and cytoprotective properties.52 An important part of their action is associated with remodelling of the pulmonary vascular bed and subsequent reduction in endothelial cell injury and hypercoaguability.53
There are two agents currently available for clinical use: epoprostenol (Flolan; Glaxo SmithKline, UK) which is licensed for use in severe primary pulmonary hypertension, and iloprost (Schering Healthcare, UK) which is not yet licensed in the UK. Both appear to be effective but the number of patients studied with iloprost is small. Epoprostenol is reconstituted in a glycine buffer (pH 10.5) and is inactivated in the circulation within five minutes.54 Iloprost has a similar molecular structure to epoprostenol and exerts its action via epoprostenol vascular endothelial receptors.55 56 It has a pH of 8.3 and a half life of > 13 minutes.57 Iloprost is also more potent than epoprostenol and only half the dose is required to produce the same long term effects. Since prostaglandins are inactivated by gastric pH, they are administered by continuous intravenous infusion using a portable infusion pump via a Hickman line.
Epoprostenol increases cardiac output and reduces pulmonary vascular resistance (PVR) during long term therapy.58 It improves measures of quality of life and exercise capacity.26 In one randomised controlled trial it also improved the survival chance of patients with PPH in modified NYHA functional classes III and IV over a three month period.26 Compared to historical controls epoprostenol improved survival when corrected for severity of disease after one year25 and after five years when survival was almost doubled (54% for epoprostenol group, 27% for controls).37 These beneficial effects may persist during long term therapy up to 10 years.17
A beneficial haemodynamic response is seen chronically in patients with no acute pulmonary vasodilator response.26 37 Those patients with a negative response to an acute vasodilator challenge have the greatest survival benefit from prostaglandin therapy.25 Patients with the most severe disease (pulmonary arterial oxygen saturation ⩽ 63%) have improved survival at one, two, and three years on therapy.25
Prostaglandin therapy doubles the time on the waiting list for lung transplantation while reducing the risk and improving the outcome of transplantation.25 59 Epoprostenol reduces the monthly mortality risk by 66% whereas lung transplanation reduces this risk by 18%.25 The need for transplantation is eliminated in some patients.59
Benefit has also been reported in pulmonary arterial hypertension caused by familial pulmonary hypertension, anorectic agents, connective tissue diseases,60 61 shunts associated with congenital heart disease,61 62 portopulmonary hypertension,61 63 chronic thromboembolic disease,61 64 sarcoidosis,61 and HIV.65
Since tolerance develops to intravenous prostaglandins the dose requirement increases over time.66 The mechanism of tolerance is poorly understood. Two dosing regimes have been reported but not compared in a trial. First, worsening symptoms are used as the main indication to increase the dose.25 66 67 Second, a more aggressive approach has been advocated where the dose is increased monthly, being uptitrated against side effects.58 61 62A comparison of two reports shows that after 17 months of therapy the dose of epoprostenol is twice as much using the aggressive regime.25 58 Chronic overdosing with epoprostenol may result in high output heart failure.68
Side effects of prostaglandin therapy include jaw pain, diarrhoea, abdominal pain, headache, flushing, arthralgia, and muscle pain. Ascites and pulmonary oedema may result from increased vascular permeability caused by these drugs. Major complications may arise from the complex delivery system, catheter infection, catheter thrombosis, and paradoxical embolism. The expected local central line infection rate is 0.22–0.68 per patient year and that of septicaemia 0–0.39 per patient year.26 58 61 62 Infection can cause worsening of PPH and may result in death. Pump or catheter malfunction has been reported up to 2.53 events per patient year.26 Symptoms recur if the infusion has to be stopped and this can result in death as a result of rebound pulmonary hypertension.
Prostaglandins may also be administered by nebuliser and have been shown to have a beneficial acute effect.20 69 Nebulised iloprost appears to be safe and produces sustained improvement in exercise capacity and haemodynamics after 12 months.70 It may be effective in severely ill patients when administered chronically.71 Randomised trial data are awaited. The high pH of epoprostenol makes it unsuitable for long term inhaled therapy.
A prostacyclin analogue, UT-15 (United Therapeutics, USA), which is administered subcutaneously has been being investigated in a double blind, placebo controlled, multicentre, randomised trial, the final results of which are awaited. UT-15 is currently available as part of a registry trial and a UK licence will be applied for in the near future.
Prostaglandin therapy is expensive. The approximate cost of the first year of treatment with epoprostenol in the UK in 1999 was typically £45 000 and iloprost £37 000. At these prices the cost of epoprostenol per quality of life adjusted year (QALY) is £127 00072 but that of iloprost is less. It is likely that the cost of therapy will fall significantly during the next year.
Failure to fund prostaglandin therapy or to respond with improved exercise tolerance over at least one month on prostaglandin therapy is an indication to consider lung transplantation. The Trent Institute for Health Services Research has recommended that prostaglandins should be administered from a limited number of centres according to agreed protocols to ensure that data on costs and outcomes are collected to inform future purchasing decisions.72
Recommendation: Patients with pulmonary hypertension which is primary, familial or caused by anorectic agents, connective tissue diseases, shunts associated with congenital heart disease, portal hypertension, sarcoidosis, HIV or chronic thromboembolic disease (either inoperable or as a bridge to pulmonary thromboendarterectomy), in modified NYHA functional classes III and IV, with a cardiac index ⩽ 2.1 l/min/m2, and/or pulmonary arterial oxygen saturation ⩽ 63% and/or right atrial pressure ⩾ 10 mm Hg should be considered for long term intravenous infusion of prostaglandins. This is regardless of whether they have any evidence of vasodilator capacity at cardiac catheterisation. Prostaglandin therapy should be considered in all patients who do not respond to conventional medical therapy before proceeding to lung transplantation. The drug should not be discontinued because of the risk of death from rebound pulmonary hypertension.
The starting dose of epoprostenol is 2 ng/kg/min and of iloprost 1 ng/kg/min. After initiation of therapy these drugs should be uptitrated over one week to the maximum tolerated dose while the patient remains in hospital. Patients should be instructed in sterile techniques for handling drugs, catheter care, preparation of dressings and drug administration. Appropriate support must be provided at home.
Conventional medical therapy should be continued after commencing prostaglandins. Calcium antagonists should be withdrawn on starting prostaglandins if there is evidence of clinical deterioration before this. The dose of prostaglandins should be increased any time the patient has a return of symptoms attributable to pulmonary hypertension.
Specialist care in a pulmonary vascular unit is required for this technically challenging treatment.
Atrial septostomy
Atrial septostomy is a palliative procedure which creates a safety valve which alleviates the high pressures to which the right heart is subjected in severe disease. In selected patients it results in an immediate fall in right ventricular end diastolic pressure and systemic oxygen saturation, and is accompanied by an increase in cardiac output.73 It may improve survival74 while awaiting lung transplantation.75 Atrial septostomy is well tolerated despite the fall in systemic oxygen saturation, but it carries a high mortality in critically ill patients.76Precise indications are uncertain owing to limited data and there are no randomised trials.
Recommendation: Atrial septostomy may be considered in severe pulmonary hypertension refractory to prostaglandin therapy particularly if it is associated with recurrent syncope. It is not indicated in the critically ill with severe right ventricular failure or in patients with impaired left ventricular function. Atrial septostomy should only be performed by physicians with experience in performing this procedure with low morbidity and in a centre with expertise in pulmonary hypertension.
SURGICAL THERAPY
Thromboendarterectomy
Recurrent or incomplete resolution of pulmonary thromboembolic events may lead to the development of pulmonary hypertension. Thrombotic material organises in the pulmonary arterial tree resulting in obstruction to flow and an increase in pulmonary vascular resistance. The prognosis of patients with chronic thromboembolic pulmonary hypertension is generally poor with a five year survival of < 10% for patients with a mean pulmonary artery pressure > 50 mm Hg.77 Diagnosis is based on the results of lung imaging (see “Aetiology”).
Surgical removal of the organised thrombotic material is achieved with a bilateral thromboendarterectomy, aiming to strip away the pulmonary arterial endothelium, commencing proximally in the main pulmonary arteries and extending out to subsegmental levels. This is done on cardiopulmonary bypass with deep hypothermic circulatory arrest. This differs from pulmonary embolectomy to remove fresh non-adherent clot.
Operative mortality in experienced hands is < 10%.78Patients with significant right ventricular dysfunction (right ventricular end diastolic pressure > 15 mm Hg) and those with significant comorbidity have the greatest risk.
Pulmonary artery pressure usually falls within 48 hours of surgery with a concomitant improvement in cardiac output. Long term results show improvement in modified NYHA functional class with most patients achieving class I or II, associated with sustained reduction in pulmonary vascular resistance and improvement in right ventricular function.78-80
Recommendation: Patients of any age with chronic proximal pulmonary thromboembolic disease should be considered for pulmonary thromboendarterectomy. Patients with coronary artery or valvar heart disease may also be considered. Significant lung disease (forced expiratory volume in one second (FEV1) < 30% predicted) is a contraindication. Patients with ventriculoatrial shunts for hydrocephalus may not be suitable because they develop distal embolic disease not amenable to surgical removal.
Lung transplantation
Lung or heart lung transplantation improves quality of life and survival in patients with pulmonary hypertension. Rapid expansion of this treatment has been limited by scarcity of donor organs and is limited to approximately 10 pulmonary hypertension patients per annum in the UK. Pulmonary hypertension accounts for 23.3% of all lung transplants in the USA.81
The choice of the transplant procedure depends on the underlying disease and on the operating team's experience. Heart lung transplantation is the longest established procedure and provides the recipient with structurally normal although denervated heart and lungs.82 The coronary–tracheal collateral circulation ensures good anastomotic healing with few early complications. Cardiac rejection is not usually seen after heart lung transplantation. Heart lung transplantation may be indicated for patients with primary pulmonary hypertension, valvar heart disease, Eisenmenger's syndrome with complex cardiac abnormalities, and complex pulmonary atresia.
Sequential single lung transplantation was developed to treat parenchymal lung disease in patients with normal hearts to avoid accelerated coronary atheroma and the effects of cardiac denervation.83 This form of lung transplantation can increase the availability of hearts for transplantation although it is recognised that use of the whole heart lung block for one recipient can be offset by the domino procedure in which the heart lung recipient's heart is transplanted into a second recipient. This may not be technically feasible in patients with pulmonary vascular disease receiving heart lung grafts. Dissection of the posterior mediastinum is not required in this procedure reducing blood loss and the potential for vagal and phrenic nerve injury. Sequential single lung transplantation may be offered to patients with primary pulmonary hypertension with essentially normal hearts, and patients with Eisenmenger's syndrome with simple cardiac abnormalities such as atrial septal defect, persistent ductus arteriosus, and ventricular septal defect which can be repaired.
Single lung transplantation is a third option, having the advantage of the most economical use of donor organs.84 85 There is an increased risk of pulmonary hypertensive crises and reperfusion oedema in the transplanted lung.84 Ventilation perfusion mismatch occurs since the vast majority of the cardiac output is to the newly transplanted lung while ventilation is more equally distributed between the two lungs. This mismatch may worsen with infection or rejection. These disadvantages have led to a move away from single lung transplantation as the procedure of choice.
Living related and unrelated donor lobar tranplantation has been performed in infants with pulmonary hypertension, but it is unlikely this technique will be applied to adults.
In summary, there are insufficient data to be categorical about which operation should be offered to an individual patient with pulmonary hypertension. The practice in the UK is in general to offer heart lung transplantation to such patients.
Survival after transplantation is 83% at one year, 70% at three years, and 54% at five years.86 The main cause of death in the long term remains obliterative bronchiolitis. Pulmonary hypertension has an adverse effect on the one year mortality following lung transplantation compared to parenchymal lung disease.87 Pretreatment with prostaglandins improves postoperative survival.59
The timing of lung transplantation is particulary important. Early transplantation may reduce total survival time and waiting too long increases the risk of death before transplantation. The course of the disease, the anticipated waiting time for transplantation, and expected survival after transplantation must be taken into account.
Recommendation: Lung or heart lung transplantation is indicated in patients with pulmonary hypertension with symptomatic progressive disease which, despite optimal medical and/or surgical treatment, leaves the patient in modified NYHA functional classes III or IV. The six minute walk test is a useful tool in the assessment of when to list patients for transplantation. Patients with a six minute walk test result of < 400 m should be considered for transplantation.5 88 Candidates should meet the internationally agreed guidelines for lung transplantation.89 Haemodynamic indications comprise a cardiac index ⩽ 2.1 l/min/m2, and/or pulmonary arterial oxygen saturation ⩽ 63%, and/or right atrial pressure ⩾ 10 mm Hg, and/or mean pulmonary artery pressure > 55 mm Hg.
- Pulmonary hypertension without congenital heart disease—All candidates should be evaluated for vasodilator therapy (including prostaglandins) and other medical or surgical interventions before transplant consideration (fig 4). Haemodynamic indices should be at the same level as for prostaglandin therapy (see above). It should be considered when treatment with epoprostenol is initiated, is failing, or is causing intolerable side effects.
- Pulmonary hypertension secondary to congenital heart disease—Since predictors of survival are least reliable in this group, the health of patients should have reached an unacceptable level from the patient's perspective or there should be clear evidence of right heart failure.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Management of prostaglandin therapy and lung transplantation.
CHILDREN WITH PRIMARY PULMONARY HYPERTENSION: DIAGNOSIS AND MANAGEMENT
The diagnostic work up of a child with pulmonary arterial hypertension is similar to that of an adult, although other more common causes of pulmonary hypertension in childhood must be excluded. These include congenital heart disease and persistent pulmonary hypertension of the newborn which has not regressed satisfactorily. Children can be extremely ill at presentation having deteriorated swiftly, but the principles of management are the same in children and adults. Extensive discussion with the parents is mandatory before embarking on any treatment regimen. Choice of therapy is based on the findings at cardiac catheterisation and the response to acute vasodilator testing, which includes study with nitric oxide. Greater reactivity is expected in children, based on both pathological and clinical data. The incidence of acute responders is 40% in children and 25% in adults.90 Hence more children than adults can be treated with chronic oral vasodilator therapy. Children who do not respond to acute vasodilator testing are treated with continuous intravenous epoprostenol, even in early childhood. They frequently need higher doses than adults and they experience the same side effects. In general, they respond better than adults. Children as young as 8 years have been treated with the subcutaneous prostacyclin analogue UT-15, but pain at the injection site is a limiting factor. Atrial septostomy can be carried out at the time of a diagnostic catheter, particularly if there is a history or suspicion of drop attacks. This avoids the risk of a second intervention and anaesthetic in a critically ill child. All require warfarin, some need diuretics, and domiciliary oxygen is helpful. Family members are screened and genetic counselling is often requested.
SUPPORTIVE CARE
Patients' association
In 2000 a patients' association was founded in the UK to provide information and support for patients and their families. More information can be obtained at <http://www.pha-uk.com>.
Contraception and pregnancy
Pregnancy is poorly tolerated and fatalities occur both during pregnancy and in the post-partum period. Combined oral contraceptives increase the risk of venous thromboembolism91 and are contraindicated in pulmonary hypertension. Data on the risks associated with progestogen only pills are limited although they may be safer.92 93 Laparascopic tubal ligation is the most reliable form of contraception and this should be undertaken at a tertiary care centre with cardiac anaesthesia and careful monitoring.
Recommendation: Women of child bearing age with pulmonary hypertension require contraceptive advice. They should be advised against becoming pregnant. Barrier methods of contraception may be used but they are not always reliable. Very low oestrogen or progestogen only pills may be used but their safety is unproven. Tubal ligation should be considered in selected patients.
Immunisation
Any chest or systemic infection may cause haemodynamic compromise with an attendant risk. Patients are at significant risk from the complications of influenza.
Recommendation: Patients should be receive a one-off pneumococcal immunisation and annual immunisation against influenza.
Care at the end of life
For some patients symptoms such as fatigue, breathlessness, nausea, abdominal bloating, and pain may be troublesome despite maximal medical therapy. The serious nature of the disease and its associated functional limitations and psychological problems may result in a need for psychological and social support.
Recommendation: Patients with inadequate symptom control should be referred for a specialist palliative care opinion. Supportive care should be provided in the community as needed. In addition specialist palliative care can offer help with holistic care of the patient and their carers.
NEW THERAPIES
There are many new treatments which are entering trials or are being investigated in prospective studies. These include new methods of delivering prostaglandins, endothelin antagonists, and sildenafil. Inhaled nitric oxide has received approval in the USA for use in persistent pulmonary hypertension of the newborn and its role in adults is being evaluated.
Disease specific recommendation
CONNECTIVE TISSUE DISEASE
Pulmonary hypertension may develop as a complication of several connective tissue diseases. The most common association is with systemic sclerosis but the occasional patient with systemic lupus erythematosis, mixed connective tissue disease or the antiphospholipid syndrome may be affected. Patients with systemic sclerosis develop pulmonary hypertension both in isolation and secondary to pulmonary fibrosis. Patients with limited cutaneous sclerosis and those with pulmonary fibrosis require careful monitoring. In patients with limited systemic sclerosis a fall in diffusing capacity in the presence of normal lung volumes may be the first indication of impending pulmonary hypertension.
Patients are symptomatic at lower levels of pulmonary artery pressure than is usual with PPH. Although 70% of patients with systemic sclerosis exhibit a positive acute vasodilator response this may not confer a survival benefit.94
Tolerance to calcium antagonists is reduced because they may worsen oesophageal motility and precipitate reflux oesophagitis and bleeding. These patients respond to epoprostenol.60 61 95 In a 12 week randomised controlled trial of epoprostenol in scleroderma, epoprostenol improved symptoms, exercise capacity, and haemodynamics although the trial was not powered to detect survival benefit.96 The presence of collagen vascular disease presents a particular problem for lung transplantation.89
Recommendation: Patients with limited cutaneous systemic sclerosis, particularly those who are anti-centromere antibody positive, should be screened by transthoracic echocardiography annually even if they have no symptoms of pulmonary hypertension. Patients with other connective tissue diseases should be screened only if they have symptoms suggestive of pulmonary hypertension.
Patients should follow the same management plan as PPH with the disease specific provisos noted above. Patients with pulmonary fibrosis may benefit from immunosuppressive therapy where active inflammation is shown on high resolution computerised tomography scans. Angiotensin converting enzyme inhibitors are commonly used to protect renal function.
CONGENITAL HEART DISEASE ASSOCIATED WITH SEVERE PULMONARY HYPERTENSION
Some types of complex congenital heart disease with pulmonary arterial hypertension are almost invariably associated with the early, rapid development of pulmonary vascular disease. The rate at which the disease develops varies according to the type of intracardiac abnormality. However, some children with simple defects, such as ventricular septal defect or patent ductus arteriosus, also develop severe disease early. They are assumed to be genetically predisposed to develop the disease but precisely why they should do so is still not clear. Surgical repair is generally undertaken during the first year of life to prevent the development of irreversible pulmonary vascular disease—that is, Eisenmenger syndrome. Pulmonary vascular resistance usually falls to normal when surgery is carried out during the first year of life. After 2 years of age, it will usually fall but not to a normal level. Children with congenital heart disease and a high pulmonary vascular resistance survive untreated for longer than patients with primary pulmonary hypertension, having better cardiac function and, depending on intracardiac anatomy, a lower right atrial pressure.97 Pulmonary venous hypertension appears to be reversible when the left sided obstructive lesion is corrected, although it may take many months to resolve.
Assessment of operability in patients with pulmonary arterial hypertension entails accurate determination of pulmonary vascular resistance and an assessment of vasoreactivity at cardiac catheterisation using nitric oxide and other vasodilators. Postoperatively, patients with an elevated resistance are at risk of developing pulmonary hypertensive crises. These potentially life threatening episodes can usually be treated successfully with nitric oxide and other measures. Some patients present with an elevated resistance later in life. After evaluation at cardiac catheterisation a lung biopsy is sometimes needed to establish the severity of the pulmonary vascular disease. The new techniques of assessing operability and of managing the postoperative patient with nitric oxide has helped identify patients who can undergo intracardiac repair at a later age with an acceptable operative risk, but operability is not synonymous with the potential reversibility of structural disease and careful patient selection is crucial. At any age, surgical repair in the presence of established disease can accelerate its progression. The patient then effectively has primary pulmonary hypertension and can be treated accordingly.
In young children, pulmonary vascular disease can progress so rapidly that the patient can die at a few months of age with severe obstructive intimal proliferation, not having had time to develop the more advanced lesions characteristic of severe, irreversible pulmonary vascular disease. But in the majority of patients the structural abnormalities in the pulmonary vasculature are the same as those in patients with primary pulmonary hypertension and develop in the same sequence. Hence it is logical to apply the same therapeutic approach to patients with this form of secondary pulmonary hypertension as are used in patients with primary pulmonary hypertension, and to try and achieve for them the same benefits of the newly evolving treatment strategies. Using an agonist such as epoprostenol which influences pulmonary vascular remodelling and has antiproliferative effects is very appealing.50-53 Unfortunately, no studies have yet evaluated the effects of chronic medical therapy on the natural history of the Eisenmenger syndrome.
Recommendation: There is no standardised management regimen for these patients and they require highly skilled medical care. Traditionally, they have been left untreated until becoming moderately or severely symptomatic and have florid evidence of Eisenmenger syndrome. Even then the treatment is generally empirical, consisting of digoxin, diuretics, and domiciliary oxygen therapy. Some are venesected when severely polycythaemic. Adults should only be venesected to relieve symptoms of hyperviscosity when the haematocrit is > 65%.98 Probably all of these patients ought to be anticoagulated with warfarin, or aspirin if they are young. Calcium channel blockers are not normally used. Symptomatic older patients with Eisenmenger syndrome can be treated with epoprostenol using the same indications as for PPH. The inhaled and oral forms of epoprostenol will be particularly helpful in treating young children. Patients in modified NYHA functional classes III or IV who are no longer responding to maximal medical or surgical treatments may need heart lung transplantation or possibly intracardiac repair with lung transplantation (see “Lung transplantation”). The decision to proceed to transplantation is based on an estimation that the intended benefit exceeds the potential risk.99 The same caveats apply to contraception and pregnancy as PPH.100
PULMONARY VENO-OCCLUSIVE DISEASE AND PULMONARY CAPILLARY HAEMANGIOMATOSIS
The clinical presentation of pulmonary veno-occlusive disease and pulmonary capillary haemangiomatosis is indistinguishable from other forms of pulmonary hypertension. High resolution CT of the lungs demonstrates characteristic appearances including centrilobular nodules, ground glass opacities and septal thickening, which are not seen in pulmonary arterial hypertension.101-103 Diagnosis may also be established by lung biopsy but this carries a significant surgical risk.104 These patients may develop life threatening pulmonary oedema with vasodilators.36 105
Recommendation: Patients with evidence of veno-occlusive disease on CT scans should not undergo an acute vasoreactivity study. These patients should be treated with conventional medical therapy with the exception of calcium antagonists and prostaglandins. Where appropriate they may be referred for lung transplantation. Atrial septostomy is a possible therapeutic option.
Outpatient follow up
Patients with pulmonary hypertension require life long monitoring in a specialist centre with the instigation of appropriate therapies as the disease evolves. Patients with pulmonary hypertension caused by hypoxia, chronic heart failure, and congenital heart disease will be followed up by other physicians and do not normally require monitoring in a specialist pulmonary hypertension centre.
Submaximal exercise testing is a useful objective assessment since exercise capacity and severity of pulmonary hypertension are correlated.26 Echocardiography may be used to monitor the effects of therapy on right ventricular function and pulmonary artery pressure.40 106 Routine monitoring should include clinical evaluation incorporating quality of life, exercise testing, chest radiography, arterial blood gases, and echocardiography.
Acute deterioration which is unexplained requires full investigation and may require cardiac catheterisation.
Acknowledgments
We are grateful to the following for reviewing this manuscript and for their helpful contributions: Dr JG Coghlan, Consultant Cardiologist, Royal Free Hospital; Professor JE Deanfield, Professor of Cardiology, Great Ormond Street for Hospital for Children; Professor TW Evans, Professor of Intensive Care Medicine, Imperial College School of Science, Technology and Medicine; Dr J Pepke-Zaba, Associate Specialist, Pulmonary Vascular Diseases Unit, Papworth Hospital; Professor PA Poole-Wilson, Professor of Cardiology, National Heart & Lung Institute, Imperial College of Science, Technology and Medicine.
References
Footnotes
-
Chairman
J Simon R Gibbs
Writing group
J Simon R Gibbs Tim W Higenbottam
Specialist reviewers
Carol Black: connective tissue diseases
Paul A Corris: lung transplantation
Sheila G Haworth: children
Keith McNeil: pulmonary thromboendarterectomy
Andrew J Peacock: medical management
Magdi H Yacoub: surgery
Publication sponsored by:
Glaxo Wellcome Medical Fellowship, and Schering AG