Article Text

Statistics from Altmetric.com
Hereditary spastic paraparesis (HSP) or the Strümpell-Lorrain syndrome is the name given to a heterogeneous group of inherited disorders in which the main clinical feature is progressive lower limb spasticity. Before the advent of molecular genetic studies into these disorders, several classifications had been proposed, based on the mode of inheritance, the age of onset of symptoms, and the presence or otherwise of additional clinical features. Families with autosomal dominant, autosomal recessive, and X-linked inheritance have been described.
Historical aspects
In 1880 Strümpell published what is considered to be the first clear description of HSP. He reported a family in which two brothers were affected by spastic paraplegia. The father was said to be “a little lame”, suggesting autosomal dominant transmission.1 Both Strümpell and Lorrain added similar cases to the literature in subsequent years.2 3 Shortly after these landmark descriptions, numerous reported cases of HSP appeared in the literature. However, Pratt, who considered the presence of additional neurological features to be incompatible with the original descriptions, labelled many of these as HSP plus syndromes.4 Early reviews of these and other cases attempted to identify “pure” cases as had been described by Strümpell. However, the definition of “pure” varied among authors.5-8 It was Harding in 1981 who, after detailed clinical evaluation of 22 families, suggested criteria for classifying HSP into pure and complicated forms which have since been adopted and are discussed below.9 Further subdivision of pure HSP was also suggested by Harding based on the age of onset of the disease. It was found that families could be separated into two groups, one with onset before 35 years (type I) and the other with onset after 35 years (type II). There seemed to be clinical differences between the groups, with the type I patients having a slow and variable course compared with the more rapidly evolving type II, in which muscle weakness, urinary symptoms, and sensory loss were more marked.9Neither of these classifications are ideal, with many families not easily fitting the criteria. As is the case in other hereditary neurodegenerative disorders, the unravelling of the molecular genetic mechanisms underlying hereditary spastic paraparesis will no doubt provide a more useful and relevant classification.
Epidemiology
The prevalence of HSP varies in different studies. Such variation is probably due to a combination of differing diagnostic criteria, variable epidemiological methodology, and geographical factors. Some studies in which similar criteria and methods were employed found the prevalance of HSP/100 000 to be 2.7 in Molise Italy, 4.3 in Valle d'Aosta Italy, and 2.0 in Portugal.10-12 These studies employed the diagnostic criteria suggested by Harding and utilised all health institutions and various health care professionals in ascertaining cases from the specific region. Poloet al in 1991 reported a higher prevalence of 9.6/100 000 in Cantabria, Spain.13 In their report only hospital records were used to ascertain cases, although many secondary cases were identified by examining all at risk relatives.
Clinical features
PURE HEREDITARY SPASTIC PARAPARESIS
Criteria which have been suggested for the diagnosis of pure HSP are included in table 1. Most patients present with difficulty walking or gait disturbance, noticed either by themselves or a relative. In those with childhood onset, a delay in walking is not uncommon. Other symptoms include stiffness of legs, urinary disturbance, and premature wear on footwear. Up to 25% of affected patients are asymptomatic, emphasising the often benign nature of the disease and the importance of careful clinical evaluation of families included in genetic studies. The age of onset can be from infancy to the eighth decade.9 The marked interfamilial variation in age of onset was one of the early pointers to genetic heterogeneity in this condition. This variation may also be partly due to difficulty in ascertaining an exact date of onset, particularly in older patients who have had the disease for decades, or may reflect other as yet unknown genetic or enviromental modifying factors.
Suggested diagnostic criteria for pure hereditary spastic paraparesis and clinical features that would alert the clinician to possible alternative diagnoses
The cardinal abnormalities on examination of patients with pure HSP include spasticity, hyperreflexia, and extensor plantar responses, with weakness of a pyramidal distribution in the lower limbs. The lower limb spasticity is the prominent finding on examination, particularly in the hamstrings, quadriceps, and ankles. This pattern of hypertonicity is responsible for the classic gait with the affected person demonstrating circumduction and toe walking. Muscle weakness when present is seen in iliopsoas, tibialis anterior and, to a lesser extent, the hamstrings. A characteristic feature of HSP, which has been stressed by several authors, is the marked discrepancy between the often severe spasticity and only mild or absent muscle weakness.8 14 This is demonstrated by the patient with HSP who is wheelchair bound due to spasticity but on manual muscle testing has normal power.
Other features which some authors have included under the umbrella of pure HSP include: mild sensory abnormalities in the lower limbs, absent ankle jerks, pes cavus, urinary symptoms, mild ataxia in the upper limbs, and mild distal muscle wasting.9 15-18 This more inclusive approach seems to be validated by the multisystem involvement suggested by paraclinical investigations and neuropathological findings. Sensory impairment is seen in 10%-65% of cases of pure HSP and is found more commonly, but not exclusively, in patients with longstanding disease. It usually consists of diminished vibration sense and, less often, diminished joint position sense in the extremities of the lower limbs.9 15 16 In most patients nerve conduction studies are normal.9 15 19 20 This suggests that the abnormalities found on examination are due to a central axonopathy rather than peripheral nerve involvement. The uncommon finding of absent ankle jerks in patients with HSP probably reflects the same process.9 16 Pes cavus is not as common a finding as was originally suggested. Harding in her series found pes cavus in only one third of patients.9 Its presence may reflect the severity or duration of the illness. Urinary sphincter disturbance occurs in up to 50% of patients. This usually manifests as a combination of urgency, hesitancy, and frequency and is more common in longstanding disease.9 15-17 21 22 Anal sphincter involvement is unusual in HSP, although Schetlenset al did report a family in which faecal urgency and incontinence were present and Schwarz had earlier described a man similarly afflicted. In both reports urinary symptoms were also present.23 24 Sexual dysfunction has only been described in one family. It occurred about 10–15 years after the onset of disease, and so the patient's reproductive capacity was not dramatically affected. The problem in affected males consisted of erectile failure with retained capacity to ejaculate.25Mild muscle wasting is well recognised although uncommon in HSP. When present it is found in distal muscles in the lower limbs, usually the small muscles of the foot and tibialis anterior. It is more common in patients who have had the disease for over 10 years.9 15If muscle wasting is prominent and present early in the course of the disease, the diagnosis should be reconsidered. Upper limb involvement is relatively uncommon and usually consists of mild hyperreflexia, which may be present early in the disease. A trace of terminal dysmetria may also be found in the upper limbs, but more florid cerebellar signs are not seen. There are occasional reports of mild distal muscle atrophy but more marked involvement of the upper limbs with spasticity, weakness, or amyotrophy is not usually seen in pure HSP.9
Important negative findings on examination are normal cranial nerve function and no evidence of corticobulbar tract involvement. The possible involvement of the autonomic nervous system in HSP has not been systematically studied, although Cartlidge and Bone, when reporting on the finding of sphincter disturbance in patients with HSP considered involvement of the autonomic nervous system in three patients and performed a battery of autonomic function tests which were normal.17
The prognosis and severity of HSP varies between families and, to a lesser extent, within the same family, although life expectancy is normal. Several authors have supported the observation by Harding that in early onset HSP (<35 years), disease progression is slow with most patients remaining ambulant through most of their lives, and with only a small proportion becoming confined to a wheelchair in elderly life. This contrasts with many cases of late onset HSP (>35 years), in whom disease progression can be rapid, with most patients losing the ability to walk in their 60s and 70s.9 16 20
COMPLICATED HEREDITARY SPASTIC PARAPARESIS
Whereas in pure HSP the spastic paraparesis is the prominent feature, in complicated HSP this is one component of a much more variable phenotype. Some types of complicated HSP are extremely rare and may have only been described in a single family. In some cases it could be argued that more than one inherited disorder may contribute to the clinical features. Hereditary spastic paraparesis has been associated with many conditions including optic atrophy, retinopathy, extrapyramidal disease, amyotrophy, dementia, ataxia, mental retardation, deafness, icthyosis, peripheral neuropathy, and epilepsy. References and brief descriptions of additional clinical features reported are given in table 2.
Clinical features which may be seen in addition to spastic paraparesis in complicated hereditary spastic paraparesis
Epilepsy associated with HSP was not mentioned in earlier reviews, perhaps because with only rare case reports of one or two affected members in a pedigree, the association could have been coincidental, given the high prevalence of epilepsy in the community. However, over the past decade several authors have published case reports where epilepsy syndromes and HSP seem to be closely associated.39 41-44 No clear pattern is found between the families where this association exists. Both autosomal dominant and recessive inheritance is seen. There is no consistent seizure type associated with HSP and myoclonic, simple partial, complex partial, and generalised epilepsy have been reported. Different types of seizures have sometimes been described in affected members within the same family. Similarly, there is variability as to when the seizures commence, with examples of seizures predating and postdating the onset of the gait disturbance. Coexisting mental retardation or cognitive impairment has often been described in these families. Epilepsy is a characteristic feature of mitochondrial diseases (for example, MERRF), a group of diseases which often manifest with diverse symptoms not unlike complicated HSP. As will be discussed later, evidence for mitochondrial involvement has been found in some autosomal recessive HSP families and it will be interesting to determine whether mitochondrial dysfunction contributes to the pathogenesis of other forms of HSP.
There are families with HSP who do seem to have involvement of the peripheral nerves. Schady and Smith reported a family with asymptomatic sensory involvement detected on clinical examination. All those within the family who underwent nerve conduction studies had decreased sensory action potentials. Three patients underwent a nerve biopsy, which showed decreased numbers of large and medium sized myelinated fibres with no evidence of demyelination, suggesting an axonal neuropathy.50 Such families represent examples of complicated HSP. Earlier reports described a severe sensory neuropathy in association with HSP, with chronic painless cutaneous ulcers and neuropathic bone resorption occurring in early childhood.51 In a less severe form trophic skin changes and ulcers on the feet develop in adult life superimposed on a longer established spastic paraparesis.52 53
Dementia has been associated with HSP in pedigrees where members may show additional features such as ataxia,65dysarthria,66 dysarthria and mild facial immobility,40 cardiac disease,32epilepsy,39 and in the Mast syndrome which consists of spastic paraplegia with dementia, dysarthria, and athetosis.62 Dementia has also been reported in both autosomal dominant and recessive families, as an isolated accompaniment to spastic paraparesis.35-38 In these families, similar patterns of cognitive deficits have been found. These include various combinations of defective recent memory, impairment of attention, poor perceptual speed, poor visuomotor coordination, and forgetfulness. In the descriptions of affected people there was an absence of features suggesting major cortical involvement such as dysphasia, agnosia, and dyscalculia. This would suggest that a subcortical dementia may characterise the complicated HSP phenotype of spastic paraplegia with dementia. It is possible that milder degrees of cognitive impairment may be more frequent than previously recognised in those affected by HSP.38 This problem has not yet been systematically evaluated. In one of the few studies which carefully evaluated cognitive function, asymptomatic impairment was demonstrated in five out of seven patients from four families with pure HSP.67It seems likely, therefore, that if patients are carefully evaluated for subclinical cognitive problems, that even pure HSP will turn out to be a multisystem disorder, with involvement outside the motor system. This phenomenon has increasingly been recognised in other apparently selective motor disorders, including motor neuron disease.68 69
Findings on investigation
Investigations are useful in excluding alternative diagnoses, but do not otherwise add to the diagnostic certainty of HSP. Central motor conduction times have been reported by several authors to show either unrecordable, or delayed responses from the lower limbs, with usually normal values from the upper limbs.70-73 Somatosensory evoked potentials have been reported to be small or absent, with the abnormality again being mainly seen in the lower limbs.20 72 74 Nerve conduction studies and EMG are normal in most cases of pure HSP.9 20 73 75 76
Analysis of CSF is usually unremarkable in HSP. However, occasional reports of abnormalities have included increased protein concentrations in complicated families and raised homocarnosine concentrations in a complicated family with spastic paraplegia, progressive mental deficiency, and retinal pigmentation.18 29 77 78
On MRI the spinal cord may appear small, but other abnormalities are not usually seen. There are occasional reports of mild to moderate atrophy of intracranial structures, particularly the corpus callosum, and also of white matter lesions in the cerebral hemispheres.15 71 79 80 However, marked atrophy or changes in white matter on MRI require thorough exclusion of other conditions.
Rapid progress is being made in the molecular genetics of HSP, which will affect the way we investigate and diagnose HSP in the future, and this is discussed later.
Differential diagnosis
Hereditary spastic paraparesis should be considered a diagnosis of exclusion. It is important to consider treatable disorders that can present in a similar manner, such as B12 deficiency, dopa responsive dystonia, and structural spinal cord disorders. Disorders, the prognosis of which differs significantly from HSP, such as multiple sclerosis and familial motor neuron disease should also be excluded. The necessity for investigations will vary depending on the individual clinical picture. Table 3 details investigations which may be useful in excluding other disorders in particular patients. Only once other diagnoses are confidently excluded can patients and their families be counselled appropriately.
Differential diagnosis of hereditary spastic paraparesis
Neuropathology
Hereditary spastic paraparesis is a disease which is compatible with a normal life expectancy, with most patients dying in old age from coincidental diseases. Given the chronicity of the condition and the paucity of treatment options, a large proportion of patients may not be under regular review by a neurologist. It is therefore not surprising that there are relatively few pathological reports in HSP. Schwarz and Lui reviewed the early pathological reports in 1952, identifying seven cases of pure HSP and added their own description of cases in 1956.81 24 Only a handful of pathological reports have been published since.80 82-84 The major neuropathological feature of pure HSP is axonal degeneration that is maximal in the terminal portions of the longest descending (corticospinal tracts) and ascending (dorsal column pathways) tracts within the spinal cord. Demyelination and gliosis can accompany the axonal loss. The most severely affected pathways are the crossed and uncrossed corticospinal tracts to the lower limbs and the fasiculus gracilis fibres from the lower limbs (figure). Involvement of spinocerebellar tracts is seen in about 50% of cases. Other less common reported findings have included decreased number of Betz cells in layer V of the motor cortex in four cases, marked neuronal loss in Clarke's column in another four cases, and one case in which the cerebellum and basal ganglia showed neuronal loss.85Dorsal root ganglia, posterior roots, and peripheral nerves are usually normal, as are anterior horn cells, although one report did demonstrate degeneration of anterior horn cells throughout the spinal cord of an affected person.24

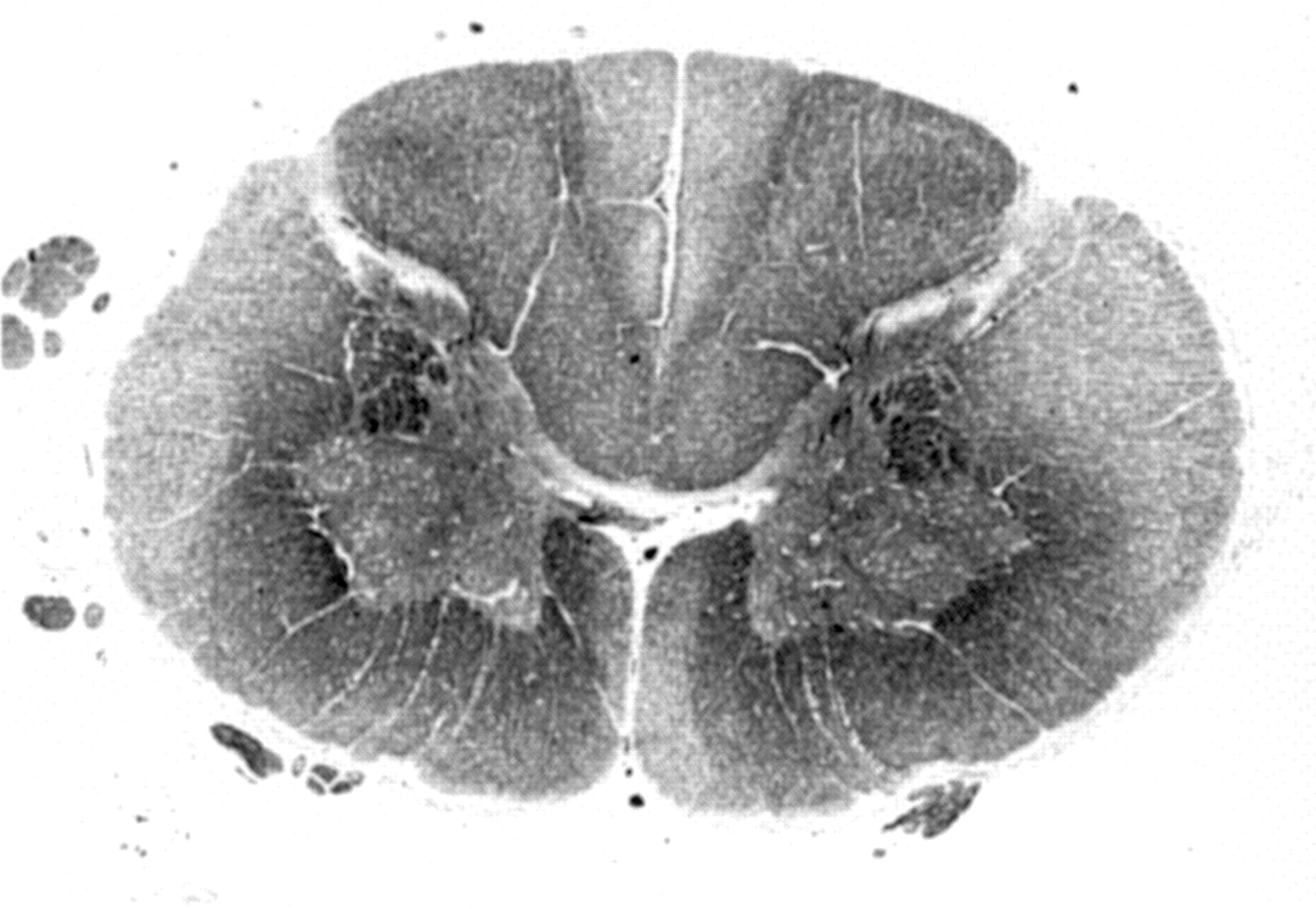{kind=link}
Midcervical spinal cord section from a case of chromosome 2 linked autosomal dominant pure HSP (SPG4), stained for myelin with Luxol fast blue. There is diffuse myelin pallor of crossed and uncrossed corticospinal tracts. In addition, the gracile fasciculi in the centre of the dorsal columns show prominent myelin pallor. Anterior horn cells are normal in number and morphology.
In the only quantitative study, the size of the pyramids in an affected person aged 57 was found to be comparable with the size in an average 2 year old. The total number of myelinated nerve fibres in the pyramid was 376 055 compared with 625 700 in a 20 year old man. In the same study the number of Betz cells was reduced to 23 652 in the left motor area compared with 34 562 in a 22 year old women. The nature of the cellular degeneration is uncertain. The reduction of Betz cells could be explained by retrograde degeneration, which can be seen when damage occurs to the spinal cord by several mechanisms. Given that the distal part of long axons in the corticospinal tracts, and sensory and spinocerebellar pathways are preferentially affected in HSP, it has been suggested that a process of “dying back” is occurring, with degeneration beginning distally and then proceeding towards the cell body.24
Pathological reports have occasionally uncovered clinically unsuspected problems such as dorsal column degeneration in patients without detectable sensory abnormalities during life. One case had documented cerebellar and basal ganglia pathology at necropsy, without any documented clinical correlate.85
Recently, new pathological changes have been described in a case of pure HSP, with autosomal dominant inheritance, linked to the chromosome 2 locus. The patient described had an early history entirely compatible with pure HSP, developing progressive spastic paraparesis from the age of 19 years. However, at the age of 72 years he developed a rapidly progressive dementing illness with several epileptic seizures in the preterminal stages. The spinal cord pathology was entirely in keeping with pure HSP. However, the brain pathology was unusual. The interesting features included a loss of neurons in the substantia nigra with associated Lewy and pale bodies, and hippocampal changes with tau immunoreactive neurofibrillary tangles. Another noteworthy feature was the presence of tau positive inclusion bodies in the limbic area and neocortex. These changes have not previously been demonstrated in HSP and also do not correspond to the pathology characteristic of other defined forms of dementia. It is possible, therefore, that these cerebral pathological features are a direct result of the expression of the abnormal SPG4 gene product.40
Genetics
The clinical heterogeneity of HSP is matched by genetic heterogeneity. Autosomal dominant, recessive, and X-linked families are well recognised with several loci described. A summary of the current genetic classification of HSP is shown in table 4.
Current genetic classification of hereditary spastic paraparesis
Autosomal dominant HSP (ADHSP)
Autosomal dominant inheritance is the commonest mode of inheritance in HSP and has been linked to loci on chromosome 2p (SPG4),38-40 71 86-94 chromosome 14q (SPG3),88-90 95-97 chromosome 15q (SPG6),98 chromosome 8q (SPG8),99chromosome10q (SPG9),64 and chromosome 12q (SPG10).100
CHROMOSOME 2P LOCUS SPG4
The chromosome 2 locus (SPG4) is by far the commonest locus to which ADHSP families are linked, accounting for about 40% of pedigrees. Recently the gene responsible for HSP at the SPG4 locus has been identified using a positional cloning strategy. The gene, spastin, is located on chromosome 2p22-p21 and is composed of 17 exons spanning a region of 90 kb.101 Spastin is a member of a group of proteins known as the ATPases associated with diverse cellular activities (AAA). The AAA proteins act in various cellular functions, including cell cycle regulation, protein degradation, organelle biogenesis, and vesicle mediated protein function.102 All of these cellular mechanisms involve the assembly and function of protein complexes and it has been proposed that AAA family members act as chaperone proteins in these complexes.103 The AAA proteins share a 230 amino acid domain which imparts the ATPase function. Outside this domain very little homology is seen, except among closely related proteins of the same AAA protein subfamily.102 Interestingly paraplegin, a member of another AAA protein subgroup, has been shown to be involved in SPG7 autosomal recessive HSP (see later).104 Spastin shares little homology with paraplegin apart from within the AAA domain and unlike paraplegin, is predicted to have a nuclear localisation. Spastin shows greater homology to a particular subclass of AAA proteins that include the yeast 26S proteasome subunits.101 Nuclear AAA proteins which exhibit homology to 26S proteasome subunits have been suggested to play an indirect part in gene regulation, inducing proteolytic activation or degradation of transcription factors.105
To date the spastin mutations identified include missense, nonsense, and splice site mutations in various exons leading to major changes in the amino acid sequence of the AAA domain. This suggests that SPG4 is caused by a loss of function implying that a threshold level of spastin protein expression is critical for axonal preservation in the corticospinal tract.101
Although most SPG4 pedigrees have pure HSP, there have been three families with complicated phenotypes also linked to this locus.38-40 In the pure HSP families linked to this locus there is marked interfamilial and intrafamilial variation in the severity and the presence or absence of features such as bladder symptoms and diminished vibration sense.
Not all the published studies comment on the severity of the motor dysfunction in affected persons and in those that do often it is not easy to directly compare different reports. There tends to be a range of severity with the majority of affected people retaining the ability to walk independently or with minimal support. At the extremes are asymptomatic patients with pyramidal signs in the lower limbs with a normal or only slightly abnormal gait and a few patients who are chairbound or bedridden.71 94
There is marked variability in the age at onset of symptoms in chromosome 2p linked families. Several published pedigrees show age at onset of symptoms ranging over 5 or more decades in different members of the same family. The mean age at onset of symptoms from the published pedigrees ranges from 12 to 43 years, demonstrating interfamilial variation also. The cause of such interfamilial and intrafamilial variation is not known. It may represent alleic heterogeneity or the presence of modifying genes or enviromental factors. Another factor may be the obvious difficulty in recognising the precise timing of onset of symptoms that begin insidiously and have often been present for many years.
In some families anticipation is reported whereby the age at onset decreases in subsequent generations.40 71 89 92This is seen in other neurodegenerative diseases—for example, Huntington's disease, dentatorubral-pallidoluysian atrophy (DRPLA), and Machado-Joseph disease (MJD). However, these diseases are caused by an abnormal CAG repeat expansion and it is the increase in length of the repeat expansion that correlates with the younger onset of disease in subsequent generations.106-108 Before the discovery of the spastin gene there were extensive searches for evidence that SPG4 was a trinucleotide repeat disorder. Both the clinical and molecular genetic evidence as to whether expanded CAG repeats were pathogenic in SPG4 were inconclusive.109-111 However, the spastin gene does not contain an abnormally expanded CAG repeat and an alternative explanation for the apparent anticipation seen in some SPG4 families must be sought. This could simply be that where anticipation is reported, this may actually represent ascertainment bias, in which the disease is noticed early in children of affected parents.
Interestingly, in three chromosome 2p linked families, HSP was associated with cognitive impairment. Webb et al described a large family with 12 affected members. One patient died of a dementing illness aged 62 and four family members with HSP were found to have dementia of subcortical type on neuropsychological testing.38 In the French family, described by Heinzleff et al, four out of seven affected family members had cognitive impairment whereas one had severe dementia with some cortical features. Three affected members with cognitive impairment also had epilepsy.39 In the family reported by White et al, the index member with HSP died with a late onset severe dementing illness with cortical and extrapyramidal features. Two other family members had memory impairment in old age, although detailed information on motor symptoms in these members was not available. One other family member with HSP had borderline learning difficulties with mild impairment in all aspects of memory. As in the other families there were family members with HSP without cognitive changes. The pathological findings in the patient with dementia in this pedigree have been discussed earlier.40
CHROMOSOME 14Q LOCUS SPG3
The first ADHSP locus to be discovered was SPG 3 on chromosome 14q and only five published families have been linked to this locus. The location of the SPG3 gene on chromosome14q11.2-q24.3 has been narrowed to a 7 cM region.88 90 95-97 Among the five families there does seem to be a trend to a relatively early age at onset of the disease, with the mean usually within the first or second decade. There has been no evidence to suggest anticipation of age at onset in successive generations. Initially it was thought that the SPG 3 phenotype may also be more severe than other subtypes of HSP. However, Huang et al and Gispertet al have published pedigrees in which the proportion of severely affected cases (those requiring a wheelchair) is not dissimilar to that seen in some SPG4 linked pedigrees.
Comparing with other loci, where it has been mentioned, there seems to be an absence of bladder or sensory involvement in the SPG 3 phenotype.
CHROMOSOME 15Q LOCUS SPG6
Only one, large North American, family has been linked to SPG6. The locus has been mapped to a ∼7.3 cM interval in the centromeric region of chromosome 15q.98 The mean age at onset was 22 years (12–35) and on the whole the phenotype was of pure HSP, although the phenotype is relatively severe. Almost one third of affected members required a wheelchair, some as early as 40 years of age. Pes cavus was almost always present in affected members, perhaps a reflection of disease severity.
CHROMOSOME 8Q LOCUS (SPG8)
Two families have been linked to the SPG8 locus on chromosome 8q24. One large North American family and more recently a British family, which enabled the genetic locus to be refined to a 3.4cM region.99 112 The mean age at onset of symptoms in the SPG8 linked pedigrees was comparable with SPG4 families. The phenotype seemed to be fairly severe, particularly in the American family, in which 10/15 affected members over 40 years required the use of a wheelchair, eight of them for more than 50% of the time.113 In both pedigrees the clinical features were typical of pure HSP with some family members having diminished vibration sense, bladder symptoms, and pes cavus. Magnetic resonance imaging demonstrated marked spinal cord atrophy in one patient with moderate symptoms from the American family linked to chromosome 8q. A reduction in cross sectional area of 50% at the T9 level, compared with age and sex matched controls, was found.113
Evidence of mitochondrial dysfunction as a mechanism of disease at this locus was not found on histochemical or biochemical analysis of a muscle biopsy taken from one affected member.113
CHROMOSOME 10Q LOCUS SPG9
One large Italian pedigree has been linked to the SPG9 locus on chromosome 10q23.3–24.2 in a region spanning 12 cM.64 The mean age at onset of symptoms varies from the first to third decade of life and the authors suggest evidence for anticipation over the three generations. The clinical phenotype is one of complicated HSP with the spastic paraparesis being associated with bilateral cataracts, gastroesophageal reflux with persistent vomiting, and distal amyotrophy secondary to an apparent axonal motor neuropathy. A pedigree with a similar complicated phenotype of congenital cataracts and spastic paraparesis has previously been described which may also prove to be due to the same gene at the SPG9 locus.114
CHROMOSOME 12Q LOCUS SPG10
The latest ADHSP locus to be discovered is SPG10 on chromosome 12q13 spanning a 9.2 cM region in a British family.100 The mean age at onset of symptoms in affected family members was younger than that seen in SPG4 pedigrees, at 10.8 (SD 9.6) years. The phenotype was one of pure HSP with a wide range of disease severity with some patients requiring a wheelchair whereas others remained asymptomatic at the age of 40 years.
Reid et al described a further pedigree in which linkage to all five known ADHSP loci had been excluded, suggesting the presence of at least one more as yet undiscovered ADHSP locus.100
Autosomal recessive HSP (ARHSP)
The recessive disease is far less common than the dominant form. Both pure and complicated phenotypes are seen. There are consanguineous families linked to three loci, SPG5 on chromosome 8p,115SPG7 on chromosome 16q116, and the recently discovered locus on chromosome 15q,117 which at present does not have an official genome database designation.
CHROMOSOME 8P LOCUS SPG5
The SPG5 locus is on chromosome 8p12-q13 and it spans an interval of 32cM. The four Tunisian families linked to SPG5 are of the pure phenotype with a mean age at onset ranging from 1–20 years. There does not seem to be a difference between the clinical features of pure ADHSP and pure ARHSP.115
CHROMOSOME 16Q LOCUS SPG7
The SPG7 locus is on chromosome 16q24.3 and at present three families have been linked to this locus. The phenotype is heterogeneous with both pure and complicated families being found. The initial family linked to SPG7 was described as pure although there were additional features, not normally seen in pure HSP, present in this family, reported by DeMichele et al, which included dysarthria.116
The protein encoded by SPG7 has been given the name paraplegin. Recently a homozygous 9.5 kb deletion in the last five exons of the paraplegin gene on chromosome 16q has been described in this original SPG7 linked family. When further unrelated patients with HSP were screened using single stranded confirmation polymorphism (SSCP) analysis, two additional frameshift mutations in the paraplegin gene were identified. One occurred in a pure ARHSP family from Italy, affected members showing a 2 bp deletion in exon 6. Another was found in a complicated ARHSP family whose additional clinical features included optic atrophy, with cortical and cerebellar atrophy seen on imaging. This mutation consisted of an insertion in the coding region of the last exon. All three SPG7 mutations lead to major abnormalities of the paraplegin protein. Paraplegin is a member of the mitochondrial metalloproteases, which are a subgroup of the AAA protein family. Other members of this subgroup include the yeast mitochondrial proteins Afg3p and Rcalp, which exhibit both proteolytic and chaperone-like activity and localise to the mitochondrial inner membrane. Targeting and localisation studies have demonstrated that the paraplegin protein localises to the mitochondrial subcellular compartment. The finding of evidence of mitochondrial dysfunction in muscle biopsy tissue from affected members of these families supports mitochondrial involvement in the disease process. The muscle abnormalities consisted of the presence of fibres negative for cytochrome oxidase and ragged red fibres which showed intense staining for succinate dehydrogenase, changes which suggest mitochondrial OXPHOS impairment.104A muscle biopsy from a patient with ARHSP linked to chromosome 8q showed no evidence of mitochondrial dysfunction, suggesting an alternative mechanism of disease in this type of HSP.113
Mitochondrial dysfunction has been identified in some neurodegenerative disorders. In diseases such as Parkinson's disease it seems that there may be direct involvement of the respiratory chain complexes. In Friedreich's ataxia and Huntington's disease the defects in OXPHOS seem to be secondary to alterations in proteins other than respiratory chain enzymes.118
Recently a paraplegin related gene AFG3L2 has been identified on chromosome 18p11.119 The predicted gene product has homology with paraplegin, the yeast mitochondrial ATPases, Afg3p and Rcalp, and AFG3L1, a related gene.119 120 AFG3L2 has also been shown to localise to the mitochondrial subcellular compartment. Further investigation is required to establish how common paraplegin defects are in the various forms of HSP and establish if AFG3L1 or AFG3L2 is also involved in causing HSP, particularly in families with a complicated phenotype, where the multisystem involvement may be reminiscent of clinical features found in other mitochondrial disorders.
This class of gene may be involved in other neurological diseases and it is interesting that AFG3L2 lies within the region of chromosome 18p to which a form of idiopathic torsion dystonia has been linked.121
CHROMOSOME 15Q
After the discovery of SPG5 and SPG7 there still remained ARHSP families unlinked to any loci. Murrilo et alhave recently uncovered what at present seems to be the commonest locus for ARHSP on chromosome 15q13–15.117 They screened eight families and found evidence of linkage to this new locus in seven.
The phenotype in these families is variable with both pure and complicated forms being described. In the pure families the age at onset was in the first decade in one and at the age of 43 years in the other, in which only one family member was affected. There was also phenotypic variation between the complicated families, although there were some similar features present in more than one family. The most interesting was atrophy or agenesis of the corpus callosum demonstrated on MRI, seen in both affected siblings from one family and in one affected member from another. In this latter member, periventricular white matter changes were also found. Both these families had similar ages at onset of disease symptoms in their second decade and mental retardation was a feature noted in one member from each family. Other associations with spastic paraplegia at this locus were mixed motor and sensory neuropathy in one family and mild cerebellar dysfunction in another two.
The association of HSP linked to chromosome 15q with agenesis of the corpus callosum and mental retardation is interesting because a gene for Andermann's syndrome or agenesis of the corpus callosum and peripheral neuropathy (ACCPN) has been shown to also have linkage to chromosome 15q.122 It may be that this form of HSP and Anderman's syndrome are allelic disorders with different mutations in the same gene giving rise to the varying phenotypes.117This is seen in X-linked HSP where different mutations in the PLP gene give rise to either a HSP phenotype or Pelizaeus-Merzbacher disease.
X-linked HSP
Although X-linked HSP is rare, its molecular genetic basis is relatively well understood.
There are two different loci identified for X-linked HSP, SPG1 and SPG2.60 123 Families with complicated HSP have been linked to both SPG1 and SPG2, whereas the pure phenotype has only been linked to SPG2.
X-LINKED SPG2 LOCUS
Complicated HSP linked to SPG2 consists of a core phenotype of spastic paraparesis, cerebellar syndrome, and mental retardation. Interfamilial variation is seen with the core features occurring to various degrees with or without additional features such as optic atrophy. Similarly, intrafamilial variation occurs.124 125 This had been reported in earlier descriptions of X-linked families.126
The gene at the SPG2 locus is the proteolipid protein (PLP) gene. Mutations, most commonly duplications, in this gene are known to cause Pelizaeus-Merzbacher disease (PMD). This is a dysmyelinating disease affecting the CNS, which consists of early onset cerebellar and pyramidal signs, with rapid progression resulting in death usually in infancy or childhood. Complicated HSP pedigrees linked to the SPG2 locus have been shown to have mutations in the PLP gene in exons 3b, 4, or 6.127 128 Therefore different mutations in the same gene can produce either a PMD phenotype or a complicated HSP phenotype. The PLP is a major myelin protein involved in both myelin maturation and maintenance in the CNS. The less severe phenotype and later development of signs in SPG2 compared with PMD is due to the fact that PLP has a smaller isoform, DM-20. In both SPG2 and PMD, PLP concentrations are reduced, whereas the concentration of DM-20 is only significantly reduced in PMD. It is thought that the two isomers have slightly different effects on myelin. The DM-20 isoform seems to be predominantly involved in myelin maturation whereas the PLP isoform, which is expressed at a later stage of development, has a role in myelin sheet compaction and maintenance. Myelin sheaths missing the PLP protein will be assembled and although they can function these eventually undergo accelerated turnover.129 These differences may explain the marked phenotypic variation between SPG2 and PMD.
A further complicated family has been found with linkage to a region containing the SPG2 locus, with no mutation identified in the PLP gene. The authors suggest this may represent evidence for a nearby third locus for X-linked HSP.130 However, in a proportion of patients with PMD no mutations have been identified.131 132 This could be due to the presence of unrecognised mutations in non-coding areas of the gene, or due to a mutation in an adjacent gene involved in either PLP gene regulation or myelin formation.
Pure X-linked HSP is very rare, with only five families described in the literature to date.123 133-136 Two of these families have been linked to the SPG2 locus.123 133 In only one of these families a single base substitution was found in exon 5 of the PLP gene, showing this pedigree of pure HSP to be allelic to complicated HSP (SPG2) and PMD.
X-LINKED LOCUS SPG1
The family with complicated HSP linked to SPG1 has marked phenotypic differences compared with the earlier pedigrees, which included the absence of cerebellar involvement, severe mental retardation, and congenital musculoskeletal abnormalities, most notably the absence of extensor hallucis longus.60 This phenotype is due to a mutation in exon 26 of the L1 cell adhesion molecule (L1CAM) gene.61 L1CAM is a transmembrame glycoprotein which is mainly expressed by neurons and Schwann cells. It plays an essential part in the development of the nervous system, being involved in axon outgrowth and pathfinding. Different mutations in this gene give rise to the syndromes of MASA (mental retardation, aphasia, shuffling gait, and adducted thumbs), X-linked hydrocephalus, and X-linked agenesis of the corpus callosum. There is much interfamilial and intrafamilial variability in families with mutations in the L1CAM gene. It is possible to have members with the various phenotypes within one family. The diseases are now referred to as being part of the clinical syndrome CRASH (corpus callosum hypoplasia, retardation, adducted thumbs, spastic paraplegia, and hydrocephalus).137
Conclusion
It is no surprise that there were early difficulties in attempting to classify HSP, which represents a heterogeneous group of diseases that at best may share a final common pathway in the neuronal degenerative process. Molecular genetic classification is now beginning to define the limits of the conditions and their genotype/phenotype correlations.
At present, mutations in four genes; L1CAM, PLP, paraplegin, and spastin have been identified as playing a part in the development of an HSP phenotype, with the identity of the genes at the other seven loci still unknown. The recent discovery of the spastin and paraplegin genes opens a new era of exciting opportunities in which we can begin to investigate the physiology of the motor system and the degenerative process it undergoes, hopefully offering opportunities for treatment. The discovery of the remaining genes and their products can be expected in the near future which will further our understanding of these diseases.
The presence of such genetic heterogeneity underlying HSP goes some way to explain the marked phenotypic variation seen between families. However, the finding of similar intrafamilial variation can only be explained by the presence of genetic modifying factors or an unknown interaction with the environment. Meyer et al demonstrated mutations in the glutamate transporter gene EAAT2, in two out of seven family members affected with pure HSP. They postulate that an interaction with the allelic variant of EAAT2 and the unknown primary genetic defect may be responsible for the phenotypic variation seen within their family.138
Despite the progress in the genetics of HSP, up to now there has been relatively little of diagnostic relevance. This may now change with the discovery of the spastin gene, mutations in which may account for up to 50% of ADHSP. When seeing a patient with possible HSP, the first step remains to exclude other diagnoses. Genetic analysis will offer the potential for precise diagnosis in cases where mutations are identified. This will ultimately offer the possibility of presymptomatic or even prenatal diagnosis in families who want to undergo such testing. However, until the full mutational range of these genes is understood, the technical feasibility of setting up such analyses remains unclear. These are multiexon genes with various mutations, so the identification of a mutation may not be particularly straightforward. The potential for precise diagnosis none the less heralds a new area of management in HSP. In the coming years further rapid progress can be expected, both in the identification of new HSP related genes and in the underlying cell biology of motor system degeneration.
Acknowledgments
PJS is supported by the Wellcome Trust as a Senior Fellow in Clinical Science.