Article Text

Statistics from Altmetric.com
The term congenital lactic acidosis (CLA) refers to a group of inborn errors of mitochondrial metabolism variably characterised by progressive neuromuscular deterioration and accumulation of lactate and hydrogen ions in blood, urine and/or cerebrospinal fluid, frequently resulting in early death.1-4 The incidence and prevalence of CLA are unknown, although it has been estimated that there are approximately 250 new cases recognised in the US per year (personal communication). Thus, with an estimated annual mortality attrition rate of 20%, at least 1000 cases exist in the general US population.
Recent diagnostic advances have allowed the biochemical or molecular identification of specific enzyme defects in the majority of infants and children with CLA. Most identifiable cases involve inherited or spontaneous mutations in the pyruvate dehydrogenase complex (PDC) or in one or more enzymes of the respiratory chain.2 4 A few cases have been reported that involve deficiencies in enzymes of the tricarboxylic acid cycle, such as fumarase, or of gluconeogenesis, such as pyruvate carboxylase (PC) or phosphoenolpyruvate carboxykinase (PEPCK). In a substantial number of patients, however, the specific biochemical defect fails to be determined by established techniques.
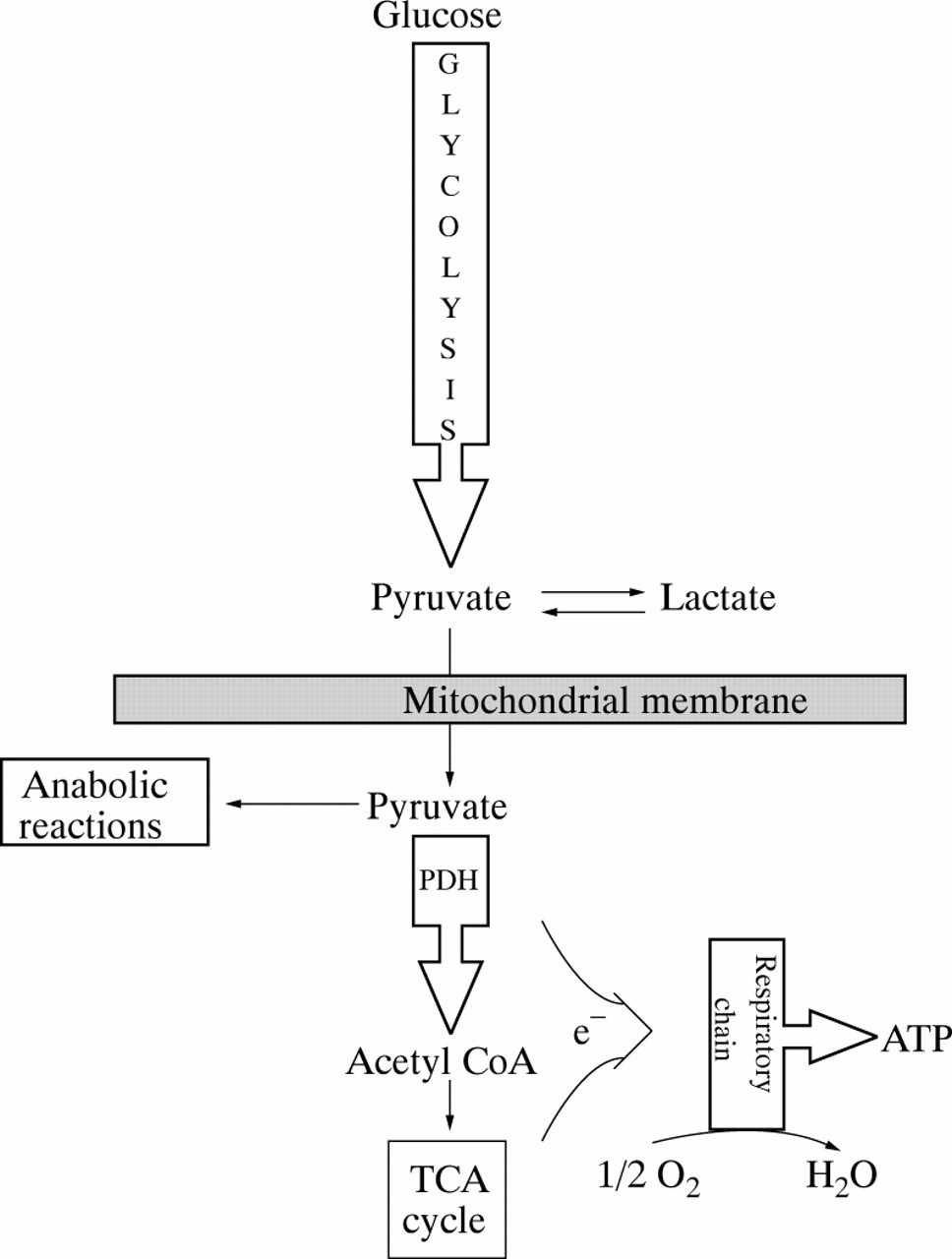
Hyperlactataemia is the defining biochemical abnormality in children with CLA and, in the absence of hypoxia, should be considered a surrogate marker for underlying failure of mitochondrial energy metabolism.5 This concept is most readily appreciated by considering mitochondrial enzyme deficiencies. Figure 1 summarises the major steps of carbohydrate oxidation in mammalian cells. Note that the oxidative fate of pyruvate is to be irreversibly decarboxylated to acetyl CoA. This reaction is catalysed by PDC, a series of linked enzymes located in the inner mitochondrial membrane (fig 2). Under aerobic conditions, the activity of PDC determines the rate at which all cells oxidise glucose, pyruvate, and lactate.
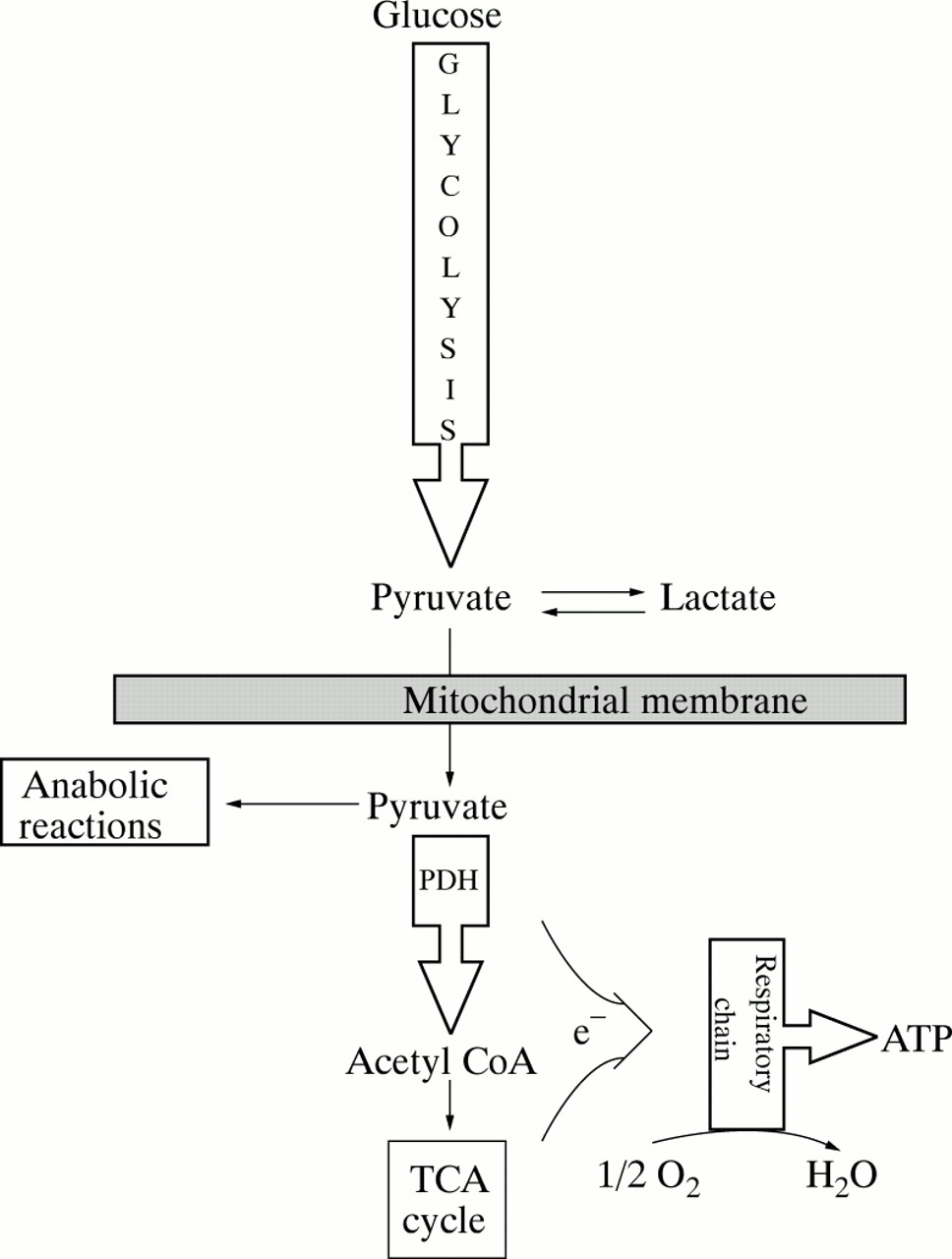
Pathways of pyruvate metabolism and oxidative phosphorylation. Pyruvate may be reduced to lactate in the cytoplasm or may be transported into the mitochondria for anabolic reactions, such as gluconeogenesis and lipogenesis, or for oxidation to acetyl CoA by the pyruvate dehydrogenase (PDH) complex (PDC). Reducing equivalents (NADH, FADH2) are generated by reactions catalysed by the PDC and the tricarboxylic acid cycle and donate electrons (e-) that enter the respiratory chain at NADH ubiquinone oxidoreductase (complex I) or at succinate ubiquinone oxidoreductase (complex II). Cytochrome c oxidase (complex IV) catalyses the reduction of molecular oxygen to water and ATP synthase (complex V) generates ATP from ADP.

The pyruvate dehydrogenase (PDH) multienzyme complex (PDC). Pyruvate is decarboxylated by the PDH subunit (E1) in the presence of thiamin pyrophosphate (TPP). The resulting hydroxyethyl-TPP complex reacts with oxidised lipoamide (LipS2), the prosthetic group of dihydrolipoamide transacetylase (E2), to form acetyl lipoamide. In turn, this intermediate reacts with reduced coenzyme A (CoASH) to yield acetyl CoA and reduced lipoamide (Lip(SH)2). The cycle of reaction is completed when reduced lipoamide is reoxidised by the flavoprotein, dihydrolipoamide dehydrogenase (E3). Finally, the reduced flavoprotein is oxidised by NAD and transfers reducing equivalents to the respiratory chain via NADH. PDC is regulated, in part, by reversible phosphorylation, in which the phosphorylated enzyme is inactive. Increases in the intramitochondrial ratios of NADH/NAD and acetyl CoA/CoA also stimulate kinase mediated phosphorylation of PDC. The drug dichloroacetate (DCA) inhibits the kinase responsible for phosphorylating PDH, thus ‘locking’ the enzyme in its unphosphorylated, catalytically active state.
PDC is regulated in part by reversible phosphorylation, in which the phosphorylated enzyme is inactive. PDC kinase catalyses the phosphorylation (inactivation) of PDC, thus inhibiting pyruvate oxidation. Persistent glycolysis without pyruvate oxidation leads to accumulation of lactate. Every molecule of lactate produced is accompanied by the generation of a hydrogen ion. However, hyperlactataemia is not necessarily associated with acidaemia, since the arterial blood pH may be normal, raised or depressed, depending on the overall clinical state and compensating respiratory mechanisms.6 Indeed, in the absence of acute illness, most patients with CLA have hyperlactataemia without acidaemia.
Mutations of subunits of the PDC system that impair the activity of the enzyme complex are likely to lead to lactate accumulation. Another probable consequence of dysfunctional PDC is cellular energy failure, due to the inability to oxidise carbohydrate and generate reducing equivalents (NADH, FADH2) required for oxidative phosphorylation, which produces ATP. This may be particularly critical to certain tissues, such as the nervous system, that rely on a high rate of oxidative glucose metabolism for normal function. It is not surprising, therefore, that developmental and degenerative neuropathology account for the cardinal clinical manifestations of PDC deficiency.1 4
Patients with defects of the electron transport chain also develop abnormal lactate accumulation and are vulnerable to cellular energy failure. Impaired electron transfer through the respiratory chain prevents oxidation of NADH and FADH2 produced by the PDC reaction and the tricarboxylic acid cycle, and ATP levels fall. The consequent rise in the intramitochondrial NADH/NAD ratio inhibits PDC activity, pyruvate oxidation decreases, and pyruvate is converted to lactate, resulting in an increased lactate/pyruvate ratio.
PDC mutations are very heterogeneous. Most arise within the coding region of the α-subunit of pyruvate dehydrogenase (the E1component), the gene for which is located on chromosome X.1 4 A few mutations also involve the gene for dihydrolipoamide dehydrogenase (the E3 component), which is located on chromosome 7. Although the majority of subunits of the various complexes of the electron transport chain are encoded by nuclear genes, virtually all mutations that have been characterised at the molecular level are the result of substitutions, deletions or rearrangements of mitochondrial DNA (mtDNA).3 mtDNA is exclusively maternally inherited. Because of its small size (16.6 kb or ∼0.001% of mammalian nuclear (n) DNA), it contains limited information. Thus, mtDNA encodes only 13 of the over 50 subunits of the respiratory chain enzyme complexes; the remainder are encoded by nDNA. Two different point mutations in mitochondrial transfer RNAs account, respectively, for at least 80% of the syndromes of mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) and myoclonic epilepsy and ragged red fibre disease (MERRF). Enzymological analyses of the electron transport chain variably show reduced complex I (NADH ubiquinone oxidoreductase) activity in MELAS individuals and both complex I and complex IV (cytochrome c oxidase) deficiencies in patients with MERRF.3
Diversity in phenotypic expression is the hallmark of mtDNA mutations.3 This is based, in large part, on a distinctive characteristic of mitochondrial genetics: the stochastic partitioning of mitochondria during cell division, a process called ‘replicative segregation’. Each mammalian mitochondrion contains up to 10 mtDNA copies per organelle. Each human cell, except mature erythrocytes, contains hundreds of mitochondria and, thus, thousands of copies of mtDNA.7 During cellular and mitochondrial division, mitochondria are randomly distributed to the daughter cells (fig 3). It follows, therefore, that replicative segregation provides a mechanism for unequal distribution of mutated mtDNA. Accordingly, the potential for marked heterogeneity in the phenotypic expression of the mutation, both among tissues of an affected individual and among individuals who harbour the same mutation, will reflect differences in the rate at which a ‘threshold’ dose of the mutation is reached.3

Replicative segregation of mitochondrial (mt) DNA. Random segregation of mitochondria during cell division ensures unequal distribution of normal (open ovals) and mutant (solid ovals) mtDNA among daughter cells. N represents the nucleus. (Reprinted, with permission, from Shoffner JM, Wallace DC. Oxidative phosphorylation diseases. Disorders of two genomes. Adv Hum Genet 1990;19:267-330.)
The variable clinical manifestations of CLA have been extensively reviewed.1-4 Patients who die during the neonatal period typically present with fulminant lactic acidosis and presumably have a profound deficiency in the activity of the affected enzyme, although this may be variably expressed in cultured fibroblasts or lymphocytes.1 2 4 In less severely affected patients, however, the magnitude of the enzyme deficit measured in cultured cells correlates rather poorly with other biochemical and clinical manifestations of the disease. This ‘fact of life’ about CLA is frustrating, but predictable, because of the variable expression of the biochemical defect and the difficulty in biopsying the most relevant tissues. Enzymological studies in skeletal muscle tissue can often be diagnostic, particularly for electron transport disorders. This is accomplished, under optimal conditions, by isolating mitochondria from a fresh biopsy specimen. Molecular genetic studies in patients with CLA offer great diagnostic potential,3 4 but their complexity emphasises both the heterogeneity in clinical presentation and course among individuals with the same mutation and the existence of different mutations among patients with the same phenotype.
Conventional treatment
Treatment of most patients with CLA has been uniquely disappointing and has been approached in a sporadic, uncontrolled manner. As recently reviewed,8 most interventions have sought to provide alternate dietary substrate fuels and/or vitamins and other cofactors that might stimulate residual enzyme activity or circumvent the enzyme defect. Carnitine, thiamin, biotin, lipoate, riboflavin, coenzyme Q, tocopherol, and vitamin K plus ascorbate have been the most commonly used agents. Administration of large doses of biotin is dramatically successful in cases of biotinidase deficiency.9 For the remaining vitamins and cofactors, however, clear benefit has been reported for only a very few individuals.
Patients with PDC deficiency do not oxidise carbohydrate efficiently, and carbohydrate containing meals may exacerbate or precipitate lactic acidosis.1 2 This has led to the use of high fat diets that induce ketosis and provide an alternative source of acetyl CoA. Ketogenic diets have been observed to reduce hyperlactataemia and to improve short term neuromuscular function in infants and children with proved PDC deficiency.8 10 The use of high fat diets seems rational on biochemical grounds and they are now generally recommended for such patients. Nevertheless, the long term benefit of this difficult nutritional treatment is limited.11
Dichloroacetate
BACKGROUND AND RATIONALE
An extensive literature on the pharmacological effects of dichloroacetate (DCA) attests to its potential safety and efficacy in treating lactic acidosis in humans.6 12 The primary mechanism is probably activation of PDC, thereby accelerating the oxidation of glucose, lactate, and pyruvate to acetyl CoA. DCA is a potent inhibitor of PDC kinase, thus ‘locking’ PDC in its unphosphorylated, catalytically active form.13 An oral dose of DCA is quickly and almost completely absorbed. The drug readily crosses the blood-brain barrier and other plasma membranes, probably via the monocarboxylate transport system that also facilitates pyruvate and lactate uptake by cells.14 In vivo and in vitro animal investigations indicate that stimulation of PDC by DCA occurs within minutes of administration in virtually all tissues, except perhaps in testes and small intestine.12
Table 1 summarises the clinical and experimental conditions in which a lactate lowering effect of DCA has been demonstrated. A controlled clinical trial of intravenous DCA versus placebo in critically ill adults with various acquired causes of lactic acidosis, such as hypotension and sepsis, showed that the drug significantly improved morbidity but did not alter survival.15 In African children with Plasmodium falciparum malaria, hyperlactataemia is one of the strongest biochemical predictors of death and is associated with a mortality up to 40%.16 DCA rapidly decreases lactic acidosis in such patients17 and significantly improves survival in a rodent model of malaria associated lactic acidosis.18 This is the first and only experimental demonstration that any intervention favourably affects survival in lactic acidosis of any aetiology.
Conditions in which the lactate lowering effect of DCA has been demonstrated
CLINICAL EXPERIENCE IN CLA
We are aware of 53 infants and children with CLA who have received oral or intravenous DCA (table 2). This information was accumulated from published reports, personal communications, and personal experience through May 1996. Most patients were male and were treated during early childhood. There was wide variation in drug dose and treatment duration. For most of the patients who received chronic DCA by mouth, the usual daily dose was 25–50 mg/kg. Higher doses, typically >100 mg/kg/day, were frequently administered to patients receiving intravenous DCA for periods of one day or a few days, which in large part accounts for the relatively high mean daily drug dose reported in table 2. It follows that the duration of DCA administration has also been highly variable. Nevertheless, 12 patients have received DCA for at least one year and three have received the drug for at least five years. Thus, the cumulative DCA treatment experience in CLA infants and children equals 41 patient years.
DCA treatment of 53 infants and children with CLA
A biochemical response to DCA administration could be determined in 49 patients and was defined as a decrease in arterial or venous blood (or plasma or serum) lactate or cerebrospinal fluid lactate of at least 20% from the pretreatment lactate level. Based on this criterion, 38 patients (76%) had a biochemical response, determined by serial measurements in blood only (27 patients) or in both blood and cerebrospinal fluid (11 patients).
In our experience of administering intravenous or oral DCA to over 200 paediatric and adult patients with acquired or congenital lactic acidosis, circulating lactate concentrations usually fall at least 20% within six hours of the initial dose, and often within two hours after single intravenous infusion of 50 mg/kg (unpublished observations).12 15 Patients who fail to achieve at least a 20% reduction in blood lactate within 24 hours of a total, daily, divided oral or intravenous dose of 25–100 mg/kg are unlikely to achieve therapeutic benefit and probably should not be retreated. In patients who do have a biochemical response to DCA, circulating lactate frequently decreases at least 30% within 24 hours and may become normal within 48 hours. The kinetics of cerebrospinal fluid lactate after DCA have been monitored in only a very few patients. In general, changes in cerebrospinal fluid and peripheral blood lactate appear to exhibit similar time courses among subjects who have a biochemical response to the drug.
Information on the subsequent clinical course of patients receiving DCA was obtained in 39 cases in sufficient detail to determine that at least 15 patients (38%) showed some clinical improvement. This number likely underestimates the true response frequency because, in most of the remaining subjects, the duration of treatment and number of doses administered were too brief or the clinical descriptions were too abbreviated to warrant conclusions about clinical efficacy.
A clinical response was defined in terms of either general physical status (for example, improved vital signs, muscle tone or exercise endurance, or decreased frequency or severity of hospitalisations) or neurological condition (for example, improved cognition or evoked potentials, decreased frequency of stroke-like episodes, or slowing or stabilisation in the rate of neurological deterioration). The following case histories are illustrative.
Case 119
A 15 year old boy with MELAS experienced visual and auditory hallucinations, delirium and violent behaviour, associated with raised blood and cerebrospinal fluid lactate concentrations. Oral administration of DCA, initially at a dose of 100 mg/kg/day, followed by a maintenance dose of 25 mg/kg/day for eight months, rapidly reduced blood and cerebrospinal fluid lactate to normal, during which time the hallucinations disappeared and his behaviour improved.
Case 2 (personal communication)20
An 11 year old girl with MELAS had persistent lactic acidosis (venous blood lactate ∼5 mmol/l), growth retardation, muscular fatigue, and frequent bouts of acid-base decompensation requiring hospitalisation. Treatment with ⩽25 mg/kg/day in two divided doses led within one week to resolution of her acidosis. Weight gain and linear growth accelerated and she grew over six inches during the initial 12 months of treatment. This patient continued to receive oral DCA for ∼ 5 years. Drug associated potential adverse effects were limited to an approximately twofold increase of serum transaminases. The patient died after an acute episode of pneumonia.
Case 3 (personal observation)
An 18 month old boy with PDC deficiency was referred to the University of Florida because of intermittent lactic acidosis (venous blood lactate ⩽10 mmol/l) and profoundly delayed neuromuscular development, characterised by generalised floppiness, inability to turn over, crawl, or sit up without assistance, and inattentiveness to auditory and visual stimuli. Twenty four hours after initiating oral DCA at a dose of 25 mg/kg twice daily, blood lactate fell 40% and the patient was notably more alert and responsive to auditory and visual stimuli. On the third day of treatment the child, for the first time, was able to rise, unaided, from recumbency and to sit upright. Improvement in behavioural and neuromuscular development continued over the ensuing two months of treatment, until the patient died of a fulminant influenza B virus infection.
Case 421
A 9 month old boy with E3 (dihydrolipoamide dehydrogenase) deficiency and recurrent hospitalisations for loss of appetite, vomiting, and lactic acidosis was treated with ⩽30 mg/kg oral DCA twice daily, for over four years (combined with thiamin and carnitine) with resolution of hyperlactataemia and maintenance of normal language development and other tests of neurological function at age 5 years.
Case 522
A 1 month old boy with cytochrome oxidase deficiency developed lactic acidosis (blood lactate 15 mmol/l), weakness, lack of ocular fixation and marked hypertrophic cardiomyopathy by echocardiography. Administration of oral DCA, at a dose of 150 mg/day in three divided doses, led to normalisation of blood lactate within 24 hours, which persisted on a maintenance drug dose of 25 mg/kg/day. When DCA was withdrawn for 12 hours, hyperlactataemia recurred. Continuous DCA administration over the ensuing nine months was associated with normalisation of cardiac size, wall thickness, and left ventricular function. Physical and neurological development (including auditory and somatosensory evoked potentials) also remained normal.
Nineteen of the 53 patients (36%) whose data comprise table 2, including each case summarised above, were treated with one or more dietary or other nutritional regimens concomitant with DCA, most often thiamin, carnitine and, in PDC deficiency, ketogenic diets. In most instances, however, these interventions preceded DCA administration and did not appear to improve the patient’s biochemical or clinical condition.
Table 3 correlates the pharmacodynamic effects of DCA in the 53 CLA patients with the reported enzymatic molecular defects, if known, or with an apparent clinical syndrome in the absence of a biochemically proved defect. Accepting the accuracy of the diagnoses, DCA was effective in reducing blood or cerebrospinal fluid lactate in most subjects, regardless of the biochemical diagnosis or clinical syndrome. Among the relatively few cases in which clinical efficacy could be evaluated, DCA administration was also associated with improvement in most diagnostic categories. Thus, these cumulative data indicate that DCA may be effective in improving morbidity in CLA due to a variety of enzymatic defects.
Efficacy of DCA in 53 patients with CLA as a function of the biochemical or clinical syndrome
A unifying theory to explain the potential effectiveness of DCA in PDC and respiratory chain defects is illustrated in fig 4. The key principle is that the amount of PDC enzyme required to cope with maximal glucose oxidation is normally high in muscle and liver but is close to rate limiting in brain, which probably explains why the central nervous system is the predominant target tissue in PDC deficiency. In an individual with partial PDC deficiency or enzyme heterozygosity, DCA would stimulate residual PDC activity by inhibiting PDC kinase. In a patient with a respiratory chain defect, muscle and liver may have an admixture of normal (‘good’) and enzyme deficient (‘bad’) mitochondria on the basis of mtDNA heteroplasmy, as previously discussed. If the proportion of ‘good’ mitochondria is adequate, activation of PDC by DCA would allow increased oxidation of lactate by peripheral tissues and produce a central→peripheral lactate gradient, with resulting egress of lactate out of brain cells, perhaps resulting in improved cerebral function. Alternatively, in females who are heterozygous for pyruvate dehydrogenase (E1alpha subunit) deficiency, cells with normal and defective E1 may be adjacent or proximal to each other, even in the brain (fig 4). In such cases, activation of E1 by DCA in the normal cells could help reduce lactate in defective cells, thereby improving overall organ function.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Potential mechanisms underlying DCA’s efficacy in pyruvate dehydrogenase (PDH) complex (PDC) or electron transport chain defects. In patients partially deficient in or heterozygous for a mutation in PDC or heteroplasmic for a mutation in a respiratory chain enzyme subunit (‘bad’ mitochondria), stimulation by DCA of PDC activity in ‘good’ mitochondria would lead to increased oxidative removal of lactate and improvement in systemic acid-base metabolism and ATP production. We are grateful to Dr Brian Robinson for conception of this diagram.
To our knowledge, chronic DCA has been generally well tolerated among patients with CLA. A male infant with PDC deficiency received up to 1500 mg/kg/day in an ultimately futile attempt to improve his acid-base disorder.28 Although no obvious drug toxicity was documented, such doses are at least an order of magnitude higher than necessary to achieve a lactate lowering effect in DCA responsive patients (see above), and should be proscribed.
Besides the mild liver abnormalities described in case 2, one 12 year old boy with PDC deficiency evaluated at the University of Florida developed a peripheral neuropathy and asymptomatic, approximately twofold increases in serum transaminases while receiving a drug dose of 37.5 mg/kg twice daily. Transaminase levels returned to normal within one month after discontinuing DCA, while clinical neuropathic signs and nerve conduction velocity studies returned to normal approximately six months after DCA was stopped. DCA was then restarted and the patient has been maintained on a dose of 12.5 mg/kg twice daily for over two years without evidence of toxicity.
A reversible peripheral neuropathy has also been produced in animals and in one adult receiving >50 mg/kg/day of DCA for several weeks.12 The neuropathy may be linked to tissue thiamin stores and may be alleviated or prevented by coadministration of thiamin in animals.45 Although a similar effect of thiamin has not been proved in patients treated with DCA, we recommend that chronic DCA administration be accompanied by a daily dose of thiamin of at least 1 mg/kg body weight.
A unifying hypothesis for CLA and its treatment
The major subtypes of CLA, while heterogeneous, nevertheless are characterised by fundamental defects in mitochondrial energetics, that is, impaired pyruvate (hence, lactate) oxidation. Therefore, it should be possible to quantify, compare, and contrast the phenotypic expressions of these fundamental defects by applying standardised clinical and biochemical tests to the CLA population in general. These variable defects of pyruvate oxidation, regardless of their precise location in the pathways of mitochondrial metabolism, may be responsive to intervention at a critical site in intermediary metabolism, specifically, at the level of PDC. These concepts are consistent with, and support, the rationale for considering DCA as a treatment for CLA. Moreover, they form the crux of the hypothesis that chronic administration of DCA to infants and children with CLA should enhance energy metabolism and, hence, cell function. Consequently, DCA may improve the quality of life of patients with CLA by (1) reducing frequency, duration, and severity of hospitalisations due to acid-base decompensation, (2) improving neurological function, and (3) stimulating linear growth.
To test these hypotheses, a randomised, placebo controlled clinical trial of DCA is now being conducted in children with biochemical and/or molecular genetic evidence of a defect in PDC or in one or more enzymes of the Krebs cycle or the respiratory chain. This is the first controlled clinical trial of any putative treatment for CLA. It also offers the first opportunity to prospectively track the natural history and course of these rare diseases in a large group of patients while they receive placebo, and should thus provide new insights into the relationships between biochemical defects and clinical phenotypes. Finally, we anticipate this investigation will provide a foundation on which to construct rigorously controlled trials of newer possible therapies for CLA as they evolve.
Acknowledgments
This work was supported by NIH grants RO1 ESO7355, P42 ESO7375 and RR 00082, by grants from the Muscular Dystrophy Association and the Children’s Miracle Network and by private donations. We gratefully acknowledge the aid of Mercy Medical Airlift and the commercial airlines that provide transportation for patients and families participating in the DCA/CLA Clinical Trial. We thank Drs Brian Robinson (University of Toronto) and Darryl DeVivo (Columbia University) for valuable discussions regarding the concept of mitochondrial energy failure in CLA and also thank Mrs Melody Riedy and Mrs Faith Clark for editorial assistance.