Article Text

Abstract
Objectives Current technologies for delivering gene testing are labour-intensive and expensive. Over the last 3 years, new high-throughput DNA sequencing techniques (next generation sequencing; NGS), with the capability to analyse multiple genes or entire genomes, have been rapidly adopted into research. This study examines the possibility of incorporating NGS into a clinical UK service context.
Methods The study applied NGS of 105 genes to 50 patients known to be affected by inherited forms of blindness in the setting of a UK National Health Service-accredited diagnostic molecular genetics laboratory. The study assessed the ability of an NGS protocol to identify likely disease-causing genetic variants when compared with current methodologies available through UK diagnostic laboratories.
Results Conventional testing is only applicable to the minority of patients with inherited retinal disease and identifies mutations in fewer than one in four of those patients tested. By contrast, the NGS assay is directed at all patients with such disorders and identifies disease-causing mutations in 50–55%, which is a dramatic increase. This includes patients with apparently ‘sporadic’ disease, and those for whom clinical management and prognosis are altered as a consequence of defining their disease at a molecular level.
Conclusions The new NGS approach delivers a step change in the diagnosis of inherited eye disease, provides precise diagnostic information and extends the possibility of targeted treatments including gene therapy. The approach represents an exemplar that illustrates the opportunity that NGS provides for broadening the availability of genetic testing. The technology will be applied to many conditions that are associated with high levels of genetic heterogeneity.
- Next generation sequencing
- retinitis pigmenttsa
- retinal dystrophy
- genetics
- diagnostics
- academic medicine
- clinical genetics
- molecular genetics
- ophthalmology
Statistics from Altmetric.com
- Next generation sequencing
- retinitis pigmenttsa
- retinal dystrophy
- genetics
- diagnostics
- academic medicine
- clinical genetics
- molecular genetics
- ophthalmology
Introduction
A decade after the completion of the sequencing of the human genome,1 we are now witnessing the dawn of a new era of genomic science. Driven by technological advances, this promises to exert a profound influence over clinical practise across all forms of medicine. Over the last 3 years, new high-throughput next generation sequencing (NGS) methods have been developed which now provide the capability to analyse, in parallel, multiple genes or even entire genomes. Aligned to complex informatics algorithms and high-performance computing, these immensely powerful sequencing strategies have been rapidly adopted by research,2 3 expanding our understanding of the genetic basis of disease at an exponential rate.
This technology has not so far been incorporated comprehensively into clinical service provision, a translational gap that has recently been highlighted.4 5 Consequently, for genetically and phenotypically heterogeneous conditions caused by mutations in one of multiple genes, current technologies for delivering gene testing still rely on conventional, sequential, gene sequencing. Labour intensive and expensive, this strategy is unable to meet even the apparently modest needs of patients with monogenic inherited disorders.
Inherited retinal disorders (such as retinitis pigmentosa (RP)) comprise a large number of conditions that cause blindness.6 Like many disease groupings, both within ophthalmology and across other specialties, RP is highly genetically heterogeneous. Genetic testing for RP has already been demonstrated to direct management, define prognosis, inform genetic counselling and help identify those for whom gene-based, or other therapies7–9 might be appropriate. However, despite such benefits, genetic testing provided in the UK is currently applied to only a small proportion of patients with RP,10 the major cause of such low uptake relating to the high cost and logistical complexity of having to consecutively screen large numbers of genes in each patient.
We have examined the use of an NGS protocol for delivering genetic testing for inherited retinal dystrophies, using this as an exemplar for analysing models of the service delivery of any genetically heterogeneous condition. We have designed, within a diagnostic setting, an NGS assay covering 105 genes (1874 individual exons) implicated in all forms of RP. Using this strategy, we demonstrate that testing can identify pathogenic mutations in over 50% of patients with RP. This represents a technological step change that has the potential to revolutionise clinical service delivery in the near term. Such a capability has important implications for clinical management, economic modelling and commissioning of services for patients with inherited disease across all medical specialities.
Methods
Identification of patients with RP
Patients diagnosed with RP were identified through the Manchester and Moorfields genetic retinal clinics. Diagnosis was based upon standard ophthalmic examination, family history and, where appropriate, supplemented with electroretinography. Fifty patients were chosen for NGS including 14 patients with autosomal dominant RP (adRP), 16 with presumed autosomal recessive RP (arRP) either on the basis of consanguinity, the presence of affected siblings or both, and 20 with sporadic RP (ie, those with no family history). Ethics approval for the study was received from the NW Research Ethics Committee (10/H1005/48). Patients referred with adRP had previously undergone conventional diagnostic testing for RP (see online supplementary table 1), while male patients referred with sporadic RP and no family history had previously undergone conventional sequencing of exon 15 of RPGR, which is mutated in X-linked RP (xlRP).11
Design of the capture array, array-based capture and re-sequencing
Targeted enrichment and sequencing were performed on 3 μg of DNA extracted from peripheral blood. Enrichment was performed with a custom designed Sure Select Target Enrichment Kit (Agilent Technologies, Santa Clara, CA,USA) for the ABI SOLiD system, following the manufacturer's protocols. The target enrichment design consisted of 105 genes (see online supplementary table 2) known to be mutated in patients with RP, Leber congenital amaurosis, macular dystrophy, achromatopsia, and Usher and Bardet-Biedl syndromes (BBS). This amounted to 1874 exons including intron/exon boundaries representing over 893 Kb of target sequence. Emulsion PCR was conducted on the resultant sample libraries. The samples were run in indexed batches of 10 on a SOLiD 4 sequencer (Life Technologies, Grand Island, NY, USA), following the manufacturer's protocols. Sequence data were mapped with SOLiD Bioscope software (Life Technologies) with the hg19 human genome as a reference.
Bioinformatics analysis
Variants were called using a combination of the Bioscope software suite and Samtools, and then filtered for those single nucleotide polymorphisms (SNPs) with ≥5× coverage. The bioinformatics analysis workflow is summarised in the online supplementary figure 1. Using Ensembl V.62, variants were initially annotated to genes, and functional consequences determined. An in-house consequence hierarchy system was used, selecting for a consequence with highest functional impact (eg, non-sense over a mis-sense) on a set of predefined RefSeq transcripts. SNPS were filtered at a novel allele depth of 18× for homozygous, and a minimum 20% allele ratio for heterozygous, and both with a minimum mean quality value (MQV) of 18 to produce high-quality SNP calls. Indels were filtered at a depth of 5×, and average end position from 10 to 40 bp.
Non-functional variants were filtered out if they were in the NHLBI Exome Sequencing Project12 in dbSNP132, unless present in The Human Gene Mutation Database (HGMD) and consequently known disease-causing mutations, or after manual assessment, as inclusion in dbSNP does not definitely exclude pathogenicity, and Genomic Evolutionary Rate Profiling (GERP) (35 species alignment) conservation score ≥0 helped to initially further prioritise variants. Sorting Intolerant From Tolerant (SIFT) Polyphen and Condel scores were reported to aid in the discrimination of variants. All potentially pathogenic variants were confirmed by Sanger sequencing. CIs were calculated using StatsDirect software. The CI stated is the approximate (Wilson) 95% mid-P CIs.
Results
Coverage of the target enrichment region and assay sensitivity
An average of 1.8 GB of DNA sequence mapped uniquely to human genome reference hg19, and of this sequence data, 92% of the 1874 targeted exons were covered at a depth of 20×. For each patient an average of 90–100 SNPs were identified (figure 1), which when filtered through the informatics pipeline described above were reduced to a small number of novel variants requiring confirmation via conventional sequencing. For those patients in this study for whom conventional testing had already been carried out, validation of the sensitivity of the NGS assay was achieved by comparing SNP detection between the two methodologies (figure 2). This demonstrated a concordance rate for SNP detection of 96% between the two technologies, and a sensitivity of the assay of 91% at depth, MQVs of 18/18, a sensitivity of 98% at 14/14 depth and MQV. The only coding region that contained SNPs identified by Sanger sequencing that were not identified using the NGS assay was RPGR ORF15, which is highly repetitive.

The average number of known, novel, insertion and deletion (in/del) variants are displayed. The SNPs are filtered for depth and mean quality value (MQV) e.g.: depth:14 and MQV:18 . The insertion and deletions are filtered by depth of ≥5

Sequence filtering of data density plot demonstrating that the majority of sequences (dark red areas) represents a sequence, which when filtered, shows high coverage (depth ≥18) and quality (minimum mean quality values ≥18). All filled triangles, representing pathogenic variants that were verified using conventional Sanger sequencing lie in the quadrant with coverage ≥18 and minimum mean quality values ≥18. A number of variants that were also confirmed by conventional Sanger sequencing (filled squares) lie outside this quadrant, suggesting that a simple diagnostic algorithm will filter out a number of significant variants.
Identification of pathogenic variants
Autosomal dominant RP
We undertook a retrospective analysis of 200 patients who had undergone the conventional adRP screen as part of routine National Health Service (NHS) diagnostic testing demonstrating that this identified a causative mutation in 44% of patients (data not shown).
We chose 14 patients at random who had no molecular diagnosis after this conventional testing strategy. The NGS assay of the 14 patients identified 9/14 (64%, 95% CI 38.7% to 83.6%) with variants that were highly likely to be pathogenic (table 1). Although a small number, if generalised, this would equate to a predicted pickup for pathogenic variants rate using the new technology of 80% of patients with adRP.
Variants identified as being highly likely to be pathogenic
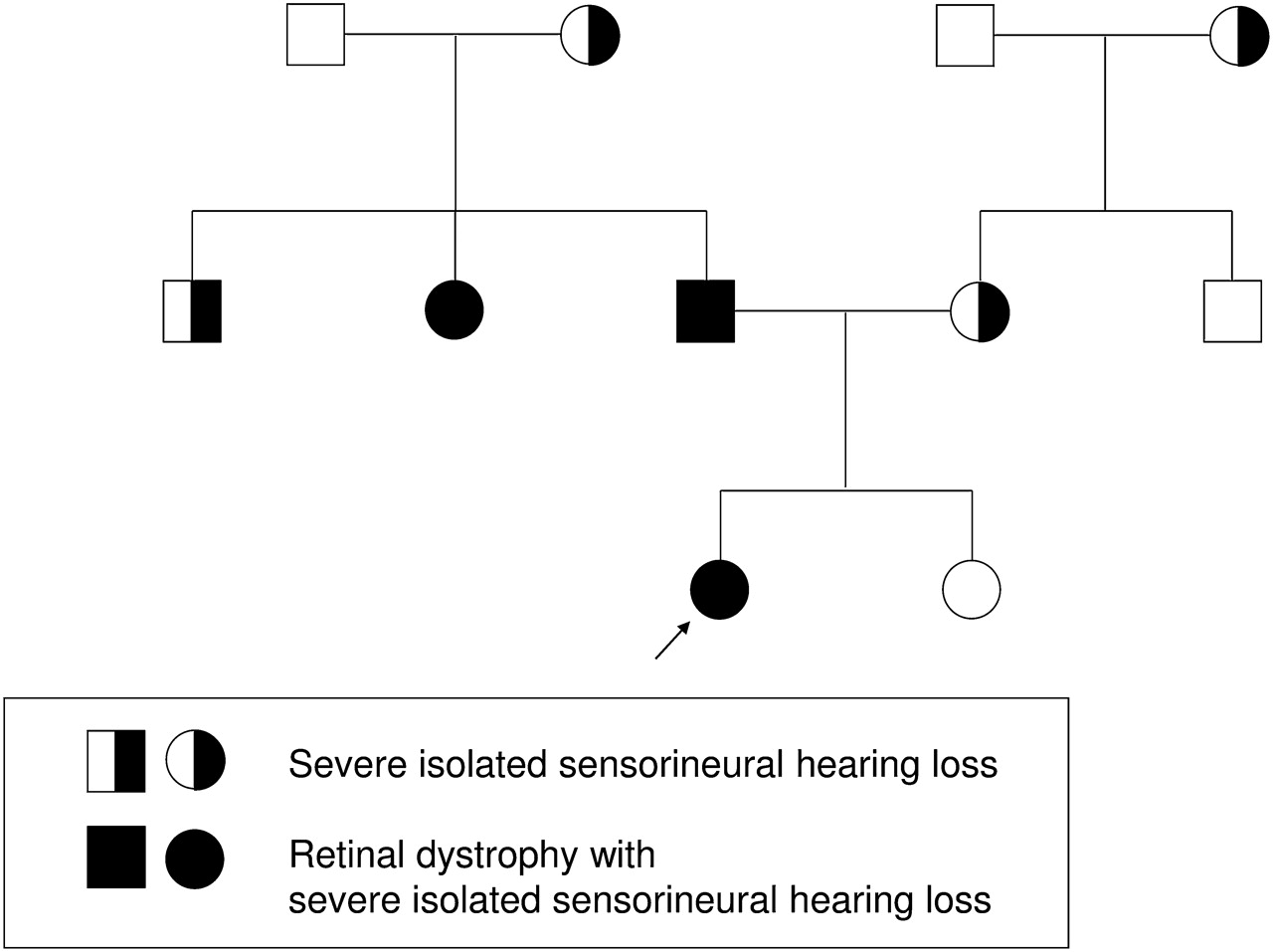
This study included patients in whom testing directly altered genetic counselling. For example, patient 3 (for pedigree, see figure 3) had a complex history of retinal degeneration and severe congenital hearing loss in multiple generations with a number of individuals with isolated hearing loss. She was found to carry compound heterozygous truncating mutations in the USH2A gene, confirming a diagnosis of the autosomal recessive Usher syndrome rather than an autosomal dominant retinal dystrophy.25

Discovering the unexpected. A family with a strong family history of autosomal dominant hearing loss and a two-generation history of retinal dystrophy had been counselled that the retinal dystrophy was likely to be autosomal dominant. The discovery of two variants in USH2A confirms a diagnosis of the recessive condition, Usher syndrome, and confirms a pseudo-dominant inheritance pattern.
Autosomal recessive RP
We selected 16 patients with presumed arRP, based either on the identification of early-onset severe retinal dystrophy or a family history of consanguinity. None had had previous testing. Of the 16 patients, we found homozygous and compound heterozygous mutations in genes associated with recessive retinal dystrophies in six patients (37%, 95% CI 18.6 to 61.35%), also listed in table 1.
As before, this group included patients for whom testing altered diagnosis and patient management.
This group included patient 10 who had a homozygous mutation in exon 12 of the BBS1 gene, previously reported to cause BBS.16 26 Classically, BBS is characterised by retinal degeneration, obesity, polydactyly and developmental delay. However, phenotypic variability is recognised and the patient will, like other patients with BBS, require screening for potential renal complications.
Another example is patient 14 who had a homozygous mutation in the SAG gene, previously reported to cause Oguchi syndrome, a form of congenital stationary night blindness.18 This suggested that the patient had a static, rather than progressive phenotype.
Sporadic RP
Twenty people with no known family history of RP were also selected. Of these 20 patients, we found six patients (30%, 95% CI 14.5% to 51.9%) with likely pathogenic variants (table 1). The average age of these 12 patients was 36 years with a range of 13–50 years at mutation detection, indicating no clear correlation between mutation detection and age at referral for this patient set. Counselling for patients with sporadic RP is strongly biased towards a diagnosis of recessive disease; however, the NGS assay allows significantly improved accuracy of diagnosis. Patients affected by RP who have no family history are most likely to have recessive disease. A major concern is defining when this may not be the case and, thereby, clarifying the risk to their offspring. This is often clinically impossible as the following two examples will illustrate. Patient 27 tested heterozygous for the previously unreported mutation in the PRPH2 gene, which is known to cause adRP. There is a 50% risk for this patient in passing on the condition to her offspring. Patient 37 is a female patient who tested heterozygous for a previously described pathogenic mutation known to cause X-linked disease, and is known to cause severe disease in men.14 She has a 50% risk of passing this mutation on to her male offspring.
Overall pickup rate of NGS diagnostic technologies for RP
Currently, the genetic testing that is available through UK testing laboratories for RD is applicable only to a minority (around one in three) of patients, specifically those with autosomal dominant and X-linked forms that account for around 35–40% of all cases. Current, conventional, Sanger sequencing strategies, therefore, largely exclude the majority of patients.
An audit of patients tested (2004–2011) demonstrates that 2104 patients have been referred for conventional testing and, assuming a prevalence of 1 in 3000–5000 for RD,27 this suggests only a small minority of patients access this genetic testing. These strategies identify pathogenic variants in 44% of adRP and 75% of xlRP, equivalent to around 24% of all cases of RD. Assuming that the proportions of the different inheritance patterns are 22:16:62 (figure 4) (adRP; xlRP; sporadic/arRP),28 29 a diagnostic portfolio combining the NGS assay with conventional sequencing of ORF15 would be estimated to identify pathogenic variants in around 50–55% of all RP patients (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Widening access to genetic testing. Currently, genetic testing available through conventional sequencing is not applicable to the majority of patients with sporadic or recessive retinal dystrophies. Next generation sequencing increases access to testing and improves the overall detection rate of mutations from 24% to >50%. AD; Autosomal dominant, AR; Autosomal recessive.
Discussion
This study has examined the feasibility of applying NGS to patients with a genetically heterogeneous disorder, using RP as an exemplar. We demonstrate that sequencing, in parallel, of 105 genes known to be mutated in all forms of inherited retinal disease enables a step change in the efficacy of testing, increasing the mutation pickup rate from 24% to 52%. Thus, it has been possible to design a pipeline that can now be used to provide a diagnostic service (http://www.mangen.co.uk/).
Not only does this strategy double the mutation pickup rate, it has the potential to dramatically increase the number of patients to whom mutation analysis can be applied. Like many genetically heterogeneous conditions, current genetic testing strategies provided within the UK are focused on a subgroup of patients only. Unlike conventional testing the novel testing platform described here extends access to molecular diagnostic evaluation.
It should be noted that this study has illuminated the fact that, using this strategy, instances of variants are thrown up that are novel and rare, but whose disease-causing status cannot be definitively declared without further study, for example, via functional or co-segregation analyses. An example is patient 1 who had a heterozygous c.148G→C (p.Gly50Arg) variant in exon 2 of the RP1 gene. This variant has not previously been described, was not found in dbSNP v.132 or the NHLBI Exome Sequencing Project, and is predicted to affect protein function in silico by SIFT, Align GVGD and POLYPHEN, but cannot be defined with certainty as pathogenic. This variant is also distinct from most other RP1 mutations causative of adRP, which are truncating and mainly found in the final exon of the gene.30
Our work does not examine the cost implications of an NGS-based diagnostic strategy. The direct costs of NGS, on a per-base assessment, are considerably lower than conventional testing. However, such theoretical savings will be strongly offset by the ability to extend access for testing, and by the increased need for genetic counselling for the patients and their families in whom pathogenic variants are identified. This point is highlighted by the individuals with isolated and sporadic (ie, those with no family history) disease who were found to have dominant and X-linked conditions, and those for whom clinical management was altered.
High-throughput NGS sequencing radically alters approaches to diagnostic testing of genetic disorders. Our diagnostic protocol illustrates a step change in the identification of pathogenic variants for a genetically heterogeneous disorder and provides a paradigm for other conditions, both ophthalmic (congenital cataract, inherited optic nerve disease) and non-ophthalmic (cardiomyopathy, cancer, ataxia, sensorineural deafness and developmental delay). This technology will extend genetic testing to large numbers of patients and has the potential to revolutionise NHS service delivery and will, as a consequence, have important implications for clinical management, economic modelling and future commissioning of services.
Acknowledgments
The authors gratefully acknowledge the financial support of Fight for Sight (Programme Grant 1801), RP Fighting Blindness (Grant GR547) and the Manchester National Institute for Health Research Biomedical Research Centre.
References
Footnotes
JO and BGM contributed equally to this research.
Funding This work was supported by Fight for Sight (Programme Grant 1801), RP Fighting Blindness (Grant GR547) and the Manchester National Institute for Health Research Biomedical Research Centre.
Competing interests None.
Ethics approval Ethics approval was provided by NW Research Ethics Committee (10/H1005/48).
Provenance and peer review Not commissioned; externally peer reviewed.