Article Text

Abstract
The ectodermal dysplasias (EDs) are a large and complex nosological group of diseases, first described by Thurnam in 1848. In the last 10 years more than 170 different pathological clinical conditions have been recognised and defined as EDs, all sharing in common anomalies of the hair, teeth, nails, and sweat glands. Many are associated with anomalies in other organs and systems and, in some conditions, with mental retardation.
The anomalies affecting the epidermis and epidermal appendages are extremely variable and clinical overlap is present among the majority of EDs. Most EDs are defined by particular clinical signs (for example, eyelid adhesion in AEC syndrome, ectrodactyly in EEC). To date, few causative genes have been identified for these diseases.
We recently reviewed genes known to be responsible for EDs in light of their molecular and biological function and proposed a new approach to EDs, integrating both molecular-genetic data and corresponding clinical findings. Based on our previous report, we now propose a clinical-genetic classification of EDs, expand it to other entities in which no causative genes have been identified based on the phenotype, and speculate on possible candidate genes suggested by associated “non-ectodermal” features.
- ectodermal dysplasia
- clinical-functional correlation
- epithelial-mesenchymal interaction
- ectodermal structural proteins
Statistics from Altmetric.com
- ectodermal dysplasia
- clinical-functional correlation
- epithelial-mesenchymal interaction
- ectodermal structural proteins
More than 170 different pathological conditions have been reported as ectodermal dysplasias,1-3 which are often associated with anomalies of other organs and systems including mental retardation.4-8 The anomalies affecting the epidermis and epidermal appendages are very variable.9 10
Previous classification
Pinheiro and Freire-Maia2 extensively reviewed EDs. They defined as ED any condition in which defects in two or more ectodermal derivatives are present. They classified the conditions according to the ectodermal structures involved and gave a number to each ectodermal derivative (hair is 1, teeth are 2, nails are 3, sweat gland function is 4); they identified 10 different subgroups for the EDs (for example, 1-2-3-4, 1-2-3). They included many case reports and personal communications in their listing of EDs, as well as conditions traditionally classified under other headings, for example dyskeratosis congenita11 and keratitis-ichthyosis-deafness (KID) syndrome12 (poikiloderma and immune defect diseases and erythrokeratodermas, respectively). Further, they did not appear to consider variability of expression and may have reported, as distinct diseases, conditions that reflect variable expression of the same pathological entity. Moreover, they included pathological conditions which, in our opinion, do not strictly fulfil the diagnostic criteria for EDs, such as conditions with secondary involvement of epidermal derivatives rather than a primary defect. We abandoned the 1-2-3-4 designation of EDs, because we believe that variable expression can render it misleading.3 The numerical system is difficult to remember and cumbersome to use.
Proposed classification
In light of what is currently known about the molecular basis and biological functions in EDs, we propose a new classification which is an attempt to integrate both molecular-genetic data and corresponding clinical findings. We basically propose two different groups, each likely to result from mutations in genes with similar function and possibly involved in the same mechanisms of regulation of development and/or pathogenesis (table 1).
Classification scheme of EDs. Italic bold entries indicate identified causative gene; Mendelian inheritance is also reported
Materials and methods
We decided only to consider those pathological conditions cited in the last revision of Pinheiro and Freire-Maia2 and also listed as OMIM (Online Mendelian Inheritance in Man) entries. We reviewed these conditions using ENTREZ in OMIM13 and searching for respective OMIM numbers. We also included five new entries obtained from ENTREZ in OMIM13using the search term “ectodermal dysplasia”, which were not included in the classification of Pinheiro and Freire-Maia.
INCLUSION/EXCLUSION CRITERIA
We strictly limited inclusion to those conditions with primary defects in at least two of the following ectodermal derivatives: hair, teeth, nails, and sweat gland function. Disorders that were reported in a single case or by personal communication were excluded, as their validity cannot be assessed and inheritance could not be established. Similarly, we did not include syndromes that have traditionally been classified under other rubrics. For example, in cardiofaciocutaneous (CFC) syndrome, hair anomalies and ulerythema ophryogenes are seen,14 but the ectodermal alterations are minor features. We did not include those pathological conditions with secondary involvement of epidermal derivatives probably resulting from a different primary defect and have classified them as “secondary EDs” (for example, Rothmund-Thomson syndrome,15trichothiodystrophies,16 and dyskeratosis congenita11).
GROUPING CRITERIA
We decided to consider first the underlying functional defect and then to try to integrate data from the clinical presentation of the related diseases. This approach is a first attempt at integration of both systems of classification and, in some ways, it may be criticised, especially when simply basing it on clinical presentation of diseases, focusing on “non-ectodermal features”, and trying to hypothesise about a possible causative gene, we arbitrarily decided to include one specific ED in one group rather than another. Nonetheless, our goal is to give both clinicians and researchers a “key” to better understanding the wide clinical variability in presentation of EDs and in dissecting “complicated” phenotypes characterised by overlap between different kinds of EDs. In the same way, we believe that this approach may help to find new candidate genes.
Group 1
The first group includes disorders in which a defect in developmental regulation and in epithelial-mesenchymal interaction can be recognised or hypothesised on the basis of an identified causative gene, its putative or proven function, and pattern of expression. There is considerable heterogeneity in clinical presentation.
The EDs included may be characterised by major ectodermal derivatives involvement. X linked anhidrotic ectodermal dysplasia (X-EDA) is the most common type of ED17 and clinically similar EDA may be inherited as autosomal dominant or autosomal recessive conditions.18 19 These are much rarer than the X linked form. All these forms are clinically indistinguishable. An altered immune response or immunodeficiency can be observed as a major clinical sign. Incontinentia pigmenti (IP) (MIM 303810), hypohidrotic ectodermal dysplasia with immunodeficiency (EDA-ID) (MIM 300291), and a newly recognised syndrome of EDA-ID with osteopetrosis and lymphoedema (OL-EDA-ID) are three conditions in this group for which a common causative gene has been identified.20-22 We included in this group other EDs with no causative gene identified in which major ectodermal derivative involvement, immunodeficiency, or functional abnormalities of the central nervous system are present. We believe that a specific functional pattern is likely to be altered in the pathogenesis of these kinds of disease.
EDs characterised by major skeletal involvement are included in this category as well. Ectrodactyly-ectodermal dysplasia-cleft lip/palate (EEC) (MIM 604292), ankyloblepharon-ectodermal dysplasia-cleft lip/palate (AEC) (MIM 106260), acro-dermato-ungual-lacrimal-tooth (ADULT) (MIM 103285), tricho-dento-osseous (TDO) (MIM 190320), and Ellis-van Creveld (EvC) (MIM 225500) syndromes are included and their causative genes have been identified.23-26 Finally, we included EDs with endocrine defects. Hypothyroidism is frequently reported in association with EDs, for example, CLPED/Fryns-Soekerman type (MIM 225040) and ANOTHER syndrome (MIM 225050).27 28 In these cases, we think that the defect in epithelial-mesenchymal interaction is likely to be caused through a different functional pattern of signalling and that candidate genes are to be searched for among nuclear proteins such as transcription factors or among regulators of gene expression.
BIOLOGICAL-FUNCTIONAL FEATURES AND IDENTIFIED CAUSATIVE GENES
An altered epithelial-mesenchymal interaction seems to be one of the most important mechanisms in the pathogenesis of EDs. During the development of skin appendages, epithelium and mesenchyme are inducers and targets of each other.3 Secreted signal molecules transmit sequential and reciprocal inductive interactions between these two structures. The interaction between ectoderm and mesoderm is sustained by the expression of specific proteins which act through different morphogenetic signalling pathways.3 Basically, two functional patterns of regulation of this interaction have been identified and recognised as possible pathogenetic mechanisms in EDs so far.
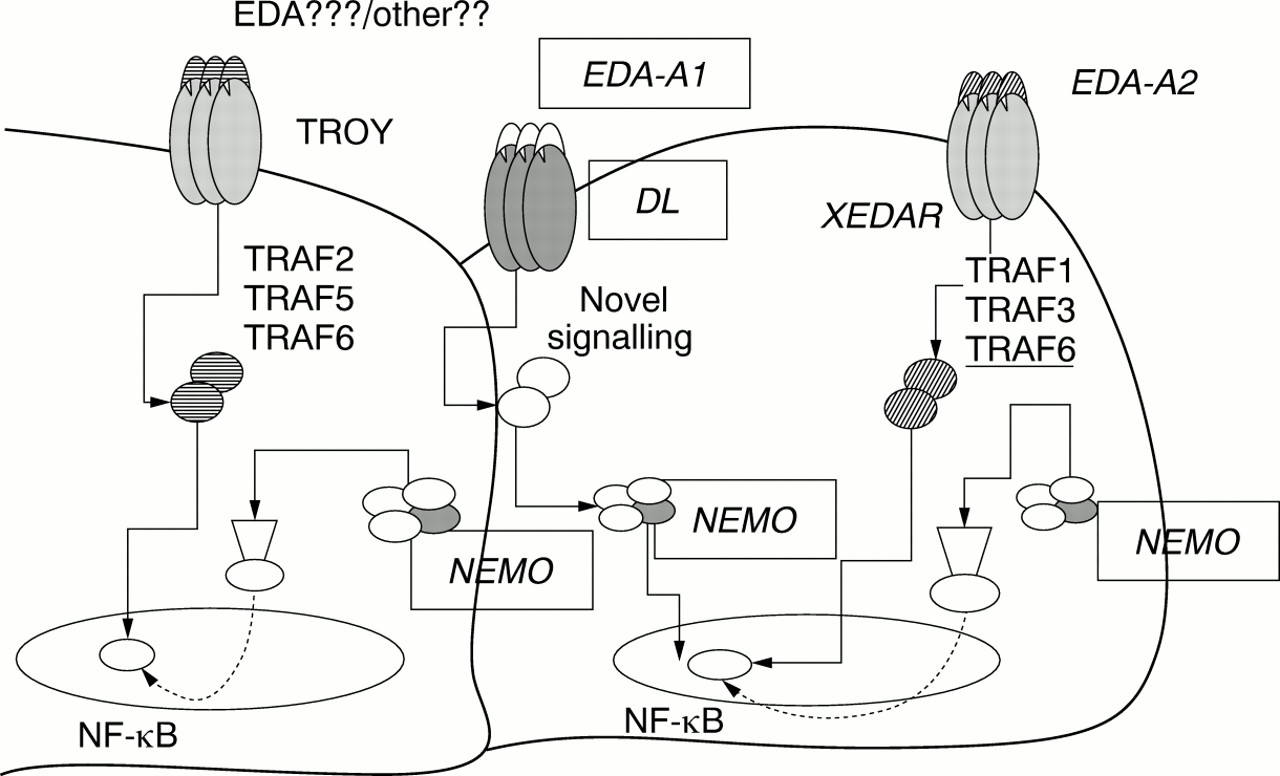
The epithelial-mesenchymal interaction may be altered if the nuclear factor kappa beta (NF-kB) regulation pattern, acting through different signalling pathways, is involved. The ectodysplasin gene isoform A1 (EDA-A1) is mutated in X linked EDA.29 The DL gene is mutated in autosomal dominant EDA and in some patients with an autosomal recessive form of EDA.30 The same clinical expression is caused by different mutations in two genes whose products physically interact with each other in a receptor ligand system.30 31 In other families with autosomal recessive EDA, linkage to the DL locus has been excluded.30 Other causative genes probably involve the same mechanism of signal transduction beyond the EDA-A1/DL binding complex, resulting in an EDA phenotype.EDA-A1 and DL are two members of the tumour necrosis factor-like (TNF-like) ligand family and the tumour necrosis factor receptor (TNFR) superfamily genes, respectively.31 32 In the mouse, theTabby (Ta) gene is homologous with human EDA. 33 Its protein product is ectodysplasin. The expression of Ta in several epithelial cell lines did not result in prominent changes in cell morphology and did not promote apoptosis, as expected of TNF-like proteins, which are critically involved in cell survival and apoptosis. On the other hand, this protein promotes cell adhesion, a function consistent with its postulated role in the epithelial-mesenchymal interface, in cell-matrix interaction, and in regulation of the development of ectodermal appendages.34 The EDA-A1/DL binding complex seems to regulate NF-kB action by enhancing the latter's activity through a specific novel signalling pathway.35 Recent findings have shown that DL triggers NF-kB through NEMO protein.22 TheNEMO gene has been found to be deleted in many cases of familial IP.20 It encodes a protein whose activity is to modulate expression of NF-kB factor.36 This action eventually regulates the expression of genes controlling apoptosis. Cells from IP patients have been found to have extreme susceptibility to apoptosis, while, clinically, IP patients may present an abnormal immune response. NEMO exon 10 mutations have been found to cause another X linked EDA with immunodeficiency (EDA-ID).21 Similarly,NEMO is mutated in patients affected with OL-EDA-ID.22 IP, EDA-ID, and OL-EDA-ID are allelic diseases with a clear genotype-phenotype correlation, loss of function mutations (large deletions) determining X linked dominant IP and hypomorphic mutations (stop codon mutation) causing X linked recessive OL-EDA-ID.22 Patients affected with EDA-ID carry mutations in the NEMO coding region, further indicating a genotype-phenotype correlation among patients withNEMO mutations. These mutations probably exert a different, milder effect than the stop codon mutation, and this would explain why EDA-ID patients have a milder clinical phenotype than OL-EDA-ID patients.22 In EDA-ID patients a residual activity of EDA-A1/DL complex is likely to exist, and this could explain the milder EDA phenotype with regard to EDA-A1/DL patients. Finally, the NEMO gene is also mutated in a peculiar hypohidrotic ectodermal dysplasia with immunodeficiency characterised by hyper IgM.37 On the basis of these results, NEMO would be a reasonable candidate gene for other EDs with immunodeficiency such as onycho-tricho dysplasia with neutropenia (MIM 258360).
A defect in epithelial-mesenchymal interaction is also observed when some regulators of transcription and/or expression of genes with functions in signalling between ectoderm and mesenchyme are mutated. p63 and DLX3 proteins are two transcription factors mutated in some cases of EEC and AEC syndrome (p63)23 24 and TDO syndrome (DLX3).25 These proteins influence the transcription of specific target genes by binding to cisacting elements located in their regulatory regions.3 TheEVC gene is mutated in some patients affected with Ellis-van Creveld syndrome.26 The function of EVC is not known, but, as previously shown for p63 and DLX3, it may also act as a regulating factor expressed in very early stages of development and may be essential for signalling pathways between ectoderm and mesenchyme.26 Its action may differentially regulate or be regulated by the expression of another gene, the collapsin response mediator protein-1 (CRMP1) (MIM 602462) located at the 3′ end of EVC and presenting a tail-tail homology with the EVC gene.38 A sort of “reciprocal” sequestering of both EVC and CRMP1 at different stages of development has been proposed as one of the mechanisms of this regulation.
Group 2
The second group includes disorders in which a structural protein defect has been found or can be inferred by specific clinical features. Group 2 EDs are characterised by heterogeneous clinical findings. Major clinical signs are hyperkeratosis, as in Clouston disease (MIM 129500),39 deafness, cleft lip/palate (CLP), as in CLPED1 syndromes (Zlotogora-Ogur/Rosselli-Guglielmetti syndrome, MIM 225000,40 and ED Margarita type syndrome, MIM 22506041), and retinal degeneration.
BIOLOGICAL-FUNCTIONAL FEATURES AND IDENTIFIED CAUSATIVE GENES
Causative genes included in this group present a specific pattern of expression, being located at the adherens junction/gap junction/apicolateral membrane domain and probably devoted to organisation of polarised apical plasma membrane domains and integrity/stability of cell membrane/cytoskeleton compartment. The underlying pathogenetic mechanism is an abnormal function of structural proteins, basically required for the correct and normal formation of ectodermal derivatives.
Connexin 30, a gap junction protein highly expressed in skin and brain, is mutated in Clouston disease.42 Gap junction proteins have been reported to be involved in cooperation between cells, growth control, and regulation of development.43 44 Interestingly, a single point mutation in the same gene causes non-syndromic autosomal dominant deafness,45 while mutations in other connexins have been found to cause syndromic palmoplantar keratoderma and deafness or erythrokeratodermia variabilis.46 47 On the basis of these data, we think that connexins are good candidate genes for EDs in which either hyperkeratosis/palmoplantar keratoderma or deafness are present.
The plakophilin 1 (PKP1) protein is altered in patients with ED/skin fragility syndrome.48 49 It is a major accessory desmosomal plaque protein. PKP1 is a member of the plakoglobulin/β-catenin/armadillo family,50 has a fundamental role in cell to cell adhesion, and may act as a linker protein between the adherens junction, desmosomes, and cornified envelope in the apico-lateral plasma membrane of epithelial cells.51 52 The biological importance of PKP1 in maintaining the integrity/stability of the cell membrane/cytoskeleton compartment is proven by the demonstration, in cells from affected patients, of defective cell to cell interaction and abnormal distribution of keratin filaments, which are disorganised and do not participate in cytoskeleton formation.48
The poliovirus receptor gene (PVRL) is mutated in some forms of ED associated with cleft lip/palate (CLPED1).53 It encodes an immunoglobulin-like membrane receptor (nectin 1), which binds a scaffold protein with PDZ domains (afadin),54 and ponsin (nectin/afadin/ponsin complex, NAP system)55 and is located in the apico-lateral membrane domain near the adherens junction compartment.56 NAP system has a central importance in maintenance of cell membrane stability and integrity through binding of afadin to actin and the cortical cytoskeleton and through binding of ponsin to vinculin and possible linkage of either these two and nectin to the cadherin-catenin system.55-58 It also has a specific role in coordinating an array of signalling and cytoskeletal proteins. Owing to the presence of PDZ domains, afadin is likely to form heteromultimeres among different structures and to be closely anchored to other plasma membrane associated proteins.59 60 Nectin 1 has a primary role in triggering an intracellular signalling cascade devoted to a correct integration of cell-cell contacts, cell morphology, and the disposition and organisation of plasma membrane domains.
We decided to include in this group EDs with retinal degeneration as a major clinical sign. Some forms of syndromic retinitis pigmentosa/retinal degeneration associated with deafness (see Usher type 1C syndrome) are the result of mutations in PDZ proteins like harmonin.61 PDZ proteins play a central role in the organisation of protein complexes in plasma membrane domains and in cell junction formation. As discussed earlier, afadin is also a PDZ protein and is involved in the formation of the NAP system and coordination and transduction of the nectin 1 signal inside the intracellular compartment.55-58 We consider afadin and other PDZ proteins good candidate genes for EDs included in group 2 and in particular for those associated with retinal degeneration.
Conclusion and future perspectives
This classification allows a different approach to the patient affected with any form of ED by using major clinical features to guide the physician to a grouping by underlying mechanisms. We believe that this approach to EDs may also help researchers to find new candidate genes using “key” clinical signs to guide molecular investigation. This kind of approach may also be applicable to some other pathological conditions we decided not to include in our classification at this first attempt, because they did not fulfil the stringent inclusion criteria, but are characterised by very similar “key signs” considered here, such as hyperkeratosis, keratoderma, or deafness. Our effort is a “work in progress” and some features discussed here may be not confirmed, in the future, by experimental findings. Nonetheless, we think that this is a useful tool in the recognition of pathological mechanisms in a confusing group of disorders.
In the EDs, different signalling pathways often converge on the same intracellular factors, or interact among them through common pathways of action, as exemplified by the role of NF-kB as a “key” molecule on which both EDA-A1/DL system and NEMO act (fig 1). Considering the augmented sensitivity to apoptosis in cells from subjects with IP, it would be interesting to test if the same phenomenon is present in both “tabby” and “dl” mice models. Recently, a novel orphan TNFR superfamily gene (TROY) has been identified, which shows high homology with DL in both coding sequence and specific pathways of expression.62 In the mouse model, Troy has been mapped near the “waved coat” (wc) locus, a mutant type characterised by skin/hair anomalies,62 so we think thatTROY is a good candidate gene for group 1 EDs. TROY also enhances NF-kB expression, but its function follows the known pathways of activation of TNF receptor associated factor (TRAF) 2, 5, and 6,63 suggesting a double way of control of NF-kB by DL/TROY receptors. We also speculate on a possible role of TROY as an additional receptor for EDA. If this was experimentally supported, EDA might activate downstream signals through two different pathways converging on the same nuclear factor (fig 1). However, TROY seems not specifically to bind EDA-A1,64 thus suggesting the existence of a novel ligand for this TNFR that has not been identified yet or, possibly, that TROY might bind a different EDA isoform. We think that another TNFR superfamily gene, the X linked ectodysplasin A2 receptor gene (XEDAR),64 is a good candidate gene for group 1 EDs. XEDAR shows a high homology with both DL and TROY TNFRs.64 XEDAR has been proven specifically to bind a second EDA isoform (EDA-A2) generated through the use of an alternative internal splice donor site of theEDA gene.64 This latter is identical to EDA-A1 except for a two amino acid residue deletion in the COOH-terminus domain. This two residue insert in EDA-A1 is thought to be on the surface of the protein in an area expected to interact with receptors. The signalling triggered by the EDA-A2/XEDAR binding system induces NF-kB activation. The cytoplasmic region of XEDAR binds TRAF1, TRAF3, and TRAF6; this last (and not other TRAFs) is specifically related to activation of NF-kB after binding with XEDAR, thus indicating that TRAF6 is probably a key adapter molecule in transducing XEDAR mediated NF-kB signalling64 (fig 1). In light of these hypotheses, the regulation of NF-kB activity in preventing apoptosis and in regulating the development of highly specific structures may be one of the most frequent mechanisms in EDs aetiopathogenesis.
{kind=link}
Intracellular pathway interactions in EDs. Group 1 molecular pathways converging on NF-kB. EDs causative genes are boxed. Some candidate genes for EDs are shown.
Some transcription factors can be considered good candidate genes for other EDs classified in group 1. As explained in our previous report,3 we think that MSX2 is a good candidate for those conditions with craniosynostosis or parietal foramina as “key signs”, such as Sensenbrenner disease (MIM 218330) and tricho-odonto-onychial dysplasia with frontal bone deficiency (MIM 275452).
Interesting molecular and functional interactions among proteins mutated in the EDs included in groups 1 and 2 and sharing as a common clinical finding cleft lip/palate may be hypothesised. CLPED1 syndromes are caused by mutations in the PVRL1 gene, which encodes a transmembrane receptor that is part of the NAP system and binds cytoskeleton and adherens junction domains.53-60 Some cases of EEC and AEC syndromes are caused by mutations inp63.23 24 The latter is likely to control transcription and expression of many genes, such asMSX1 and SHH.23 Interestingly, mutations in these genes have been proven to cause different types of CLP associated, in the case of MSX1, with defects in epidermal derivatives.65 MSX1 is supposed to interact physically with another group 1 protein, DLX3, mutated in patients affected with tricho-dento-osseous syndrome. DLX3 is a potent transactivator of some cytoskeleton protein coding genes, such as profilaggrin.66-68 It would be interesting to test p63 or other transcription factors' activities in enhancing or regulating the expression of ectodermal structural proteins. Some other EDs show overlapping phenotypes between group 1 and group 2, such as EEM syndrome, in which both ectrodactyly and macular degeneration are present.69 Again, it is intriguing to hypothesise that this complex phenotype could result from somewhat altered regulation on PDZ/cytoskeleton protein transcription by p63, even directly or mediated by other(s) factor(s). Finally, we decided arbitrarily to include Rapp-Hodgkin disease in group 1 and preliminarily to consider it as the result of a defect of epithelial-mesenchymal interaction by referring to the description of an EEC child born to a mother with clinical findings characteristic of Rapp-Hodgkin disease.70 It would be possible to consider EEC/AEC and Rapp-Hodgkin disease as variable clinical expression of the same pathological entity. Although there is no evidence ofp63 mutations in patients affected with Rapp-Hodgkin disease, other genes could be involved in the pathogenesis of the EEC/AEC/Rapp-Hodgkin disease clinical spectrum (genetic heterogeneity). Another genetic locus in 7q, named EEC1, has been suggested71 by linkage analysis studies. Two members of the DLX family, named DLX5 andDLX6, are mapped in this region. Again, complex physical or functional interactions among different proteins (in the specific case of p63 and members of the DLX family) could be hypothesised in the pathogenesis of the EEC phenotype.
Summary
The proposed classification is a great advance in understanding the biological mechanisms of EDs pathogenesis. The clinical revision of EDs using the stringent criteria described here led us to exclude single case reports and some very well known described phenotypes which do not strictly fulfil the classical definition of ED.
This first attempt at genotype-phenotype correlation in EDs has taught us that both clinical and molecular data can help in defining each single patient. Molecular interactions and pathological mechanisms can explain the many clinical signs, variability in severity, associated malformations, and overlap seen in some ED patients. Conversely, the accurate definition of “key signs” can aid researchers in an attempt to find new candidate ED genes.
Acknowledgments
We wish to thank Professor Virginia P Sybert for helpful discussion and for critical reading of the manuscript.