Article Text

Abstract
A clinical study of patients on the North West Regional Genetic Register with neurofibromatosis type 1 (NF1) identified 523 affected cases from 304 families. In those for whom relevant information was available, 86.7% (383 of 442) had more than six café au lait patches, 83.8% (310 of 370) had axillary freckling, 42.3% (151 of 357) had inguinal freckling, and 63% (157 of 249) had Lisch nodules. Cutaneous neurofibromas were present in 59.4% (217 of 365) and 45.5% (150 of 330) were noted to have subcutaneous tumours. Plexiform neurofibromas were present in 15.3% (80 of 523).
A positive family history of NF1 was found in 71.2% (327 of 459) and 28.8% (132 of 459) of affected patients were considered to be the result of a new mutation. Learning difficulties of varying severity occurred in 62% (186 of 300).
CNS tumours associated with NF1 were reported in 9.4% (49) of patients, optic gliomas occurring in 25 of these, 4.8% of patients. Some degree of scoliosis was reported for 11.7% (61), 1.9% (10) had pseudoarthrosis, 4.3% (23) had epilepsy, and 2.1% (11) had spinal neurofibromas.
Actuarial analyses were carried out for both optic glioma and malignant nerve sheath tumours and the data are presented.
- neurofibromatosis type 1
- optic glioma
- malignancy
- Lisch nodules
Statistics from Altmetric.com
Neurofibromatosis type 1 is a dominantly inherited disorder which occurs with a frequency of between 1 in 3000 and 1 in 4000.1 It is characterised by the presence of café au lait spots, peripheral neurofibromas, Lisch nodules, axillary freckling, skeletal dysplasia, and optic gliomas. The diagnosis of NF1 is based on two or more of the features listed in table 1, provided that they cannot be accounted for by any other disease.2The gene for NF1 was localised to the long arm of chromosome 17 in 19873 and has since been cloned.4-6
Diagnostic criteria for type 1 neurofibromatosis. Two or more of the following criteria are required for diagnosis
Approximately 50% of cases with NF1 represent new mutations and there may be more mutable areas of the gene.7 Penetrance in affected subjects is virtually 100% by the age of 5 years.8 9 Expression of the disease is highly variable, both between and within families. The interfamilial variability has been suggested to be the result of different mutations within the NF1 gene,1 but variation within families has pointed to the possible role of environmental alteration of gene expression or a role for modifying genes.10-12
Prenatal and presymptomatic diagnosis of NF1 is possible with a high degree of accuracy in families with more than one affected subject through linkage analysis.13 However, for the large number of families in which there is only one affected subject, it is necessary to identify the specific mutation within the NF1 gene. Unfortunately, the mutation detection rate is low.14Intrafamilial variability may make reproductive decisions difficult in families where prenatal diagnosis would be possible.15
It is because of this great variability in phenotype that population studies such as those carried out by Huson et al 16 are of great value in counselling families with NF1. A genetic register was established for patients and families with NF1 in the north west region in 1989. The following data were obtained using patient information from the register and reflect the features and complications of this condition.
Patients and methods
PATIENT ASCERTAINMENT
The genetic register for the north west region of England (population 4 million), based at the Regional Genetic Service at St Mary’s Hospital, Manchester, receives referrals from all specialties. When it was first established, patients already known to the department were contacted and offered inclusion on the register. To ascertain proactively more patients, paediatricians, neurologists, and dermatologists were informed of the service and invited to refer patients. Subsequent awareness of the register has prompted referrals by specialists in many clinical fields. New referrals continue to be made to the department and are often from general practitioners and paediatricians although may occur from many specialties. Some patients present with the features of NF1 while others are diagnosed coincidentally following examination for another condition. Any investigations which are considered clinically appropriate would be instigated at the genetic clinic.17 First degree relatives of patients who have been diagnosed elsewhere may also be referred for assessment. The service for diagnosis, counselling, support, and long term follow up is extended to all first degree relatives of affected cases. Parents of newly diagnosed children are examined for any features of NF1 to ensure an accurate recurrence risk can be given. An annual review appointment is offered for both health screening and counselling. If declined, a form to update the family details is also sent and a stamped envelope included. The department remains a point of contact for any intervening problems. Patients are included in the register only if they give their consent.
ANALYSIS OF DISEASE FEATURES
All subjects referred are first examined by doctors within the two Departments of Medical Genetics in the region and the diagnosis of NF1 is made on the basis of the National Institutes of Health Consensus Criteria.2
Cutaneous neurofibroma was used to describe the superficial, soft, greyish tumour which is well circumscribed. Subcutaneous neurofibroma referred to the discrete tumours which do not merge with the surrounding tissue. Plexiform tumours comprised large, diffuse, subcutaneous tumours which may be associated with local overgrowth.
Between May 1993 and March 1996 all new referrals and review subjects had a detailed form completed at the time of their visit. Forms for patients previously referred were completed retrospectively from clinic notes. Details of all affected subjects were transferred to a computer database.
ACTUARIAL ANALYSIS
The possibility of developing a tumour is often a major concern to patients when a personal or family diagnosis of NF1 is made. In order to quantify this risk better, we undertook actuarial analysis of the data on optic nerve gliomas and malignant nerve sheath tumours. We defined “time until an event” as the time between the date of diagnosis for a particular tumour and the date of birth. Cases were classed as censored if the patient reached the date at the end of the study free from the particular tumour, the patient died from another cause before the date of the end of the study, or if there was no follow up date for the patient but he/she was interviewed and recorded as having another type of tumour at a specific date. The reasoning being that if the patient/family were asked at that appointment if any new tumours had occurred, it would have been stated.
On the 31 March 1996, details from 523 patients affected with NF1 were available for analysis. This included index cases and relatives.
Results
Five hundred and twenty-three patients from 304 families were identified as having NF1 and 517 of the patients were still living in March 1996. These included 249 males and 274 females with a median age of 19 years (interquartile range=24 years, range 0 to 74). This equates to a diagnostic prevalence of 1 in 7650.
FAMILY HISTORY AND PEDIGREES
Three hundred and twenty-seven (71.2%) patients in the study had an affected first degree relative (parent or sib or both), with 132 (28.8%) having no positive family history for NF1. No information was available for 64 patients, four of whom were adopted. Of the 327 subjects with an affected relative, 122 had an affected father, 191 had an affected mother, and 179 had an affected sib, including numerous cases of more than one affected first degree relative.
Affected girls had been born to solely affected mothers in 108 (56.5%) cases, and affected boys to solely affected mothers in 83 (43.5%) cases. Sixty-two affected girls (50.8%) and 60 affected boys (49.2%) were born to solely affected fathers and three affected girls and one affected boy to parents who were both affected by NF1. Molecular analysis is not yet complete in this family.
Segmental NF1 was present in four patients, one of whom had a child with unequivocal NF1. (This case had been reported elsewhere.1) These cases were included in the study as they satisfied the NIH criteria for the diagnosis of NF1.
THE FREQUENCY OF MAJOR DIAGNOSTIC FEATURES OF NF1
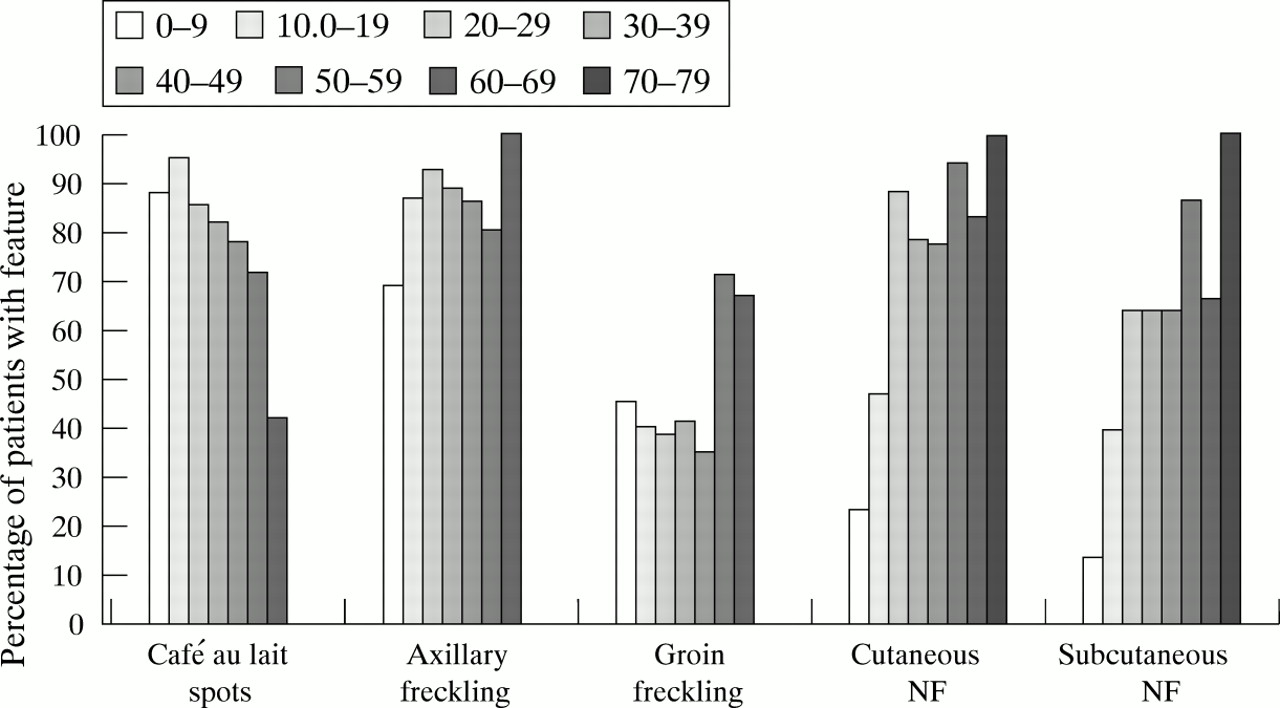
Fig 1 shows the overall percentage of patients with diagnostic features in the different age groups. The total number of patients examined for each of the various features of the condition in each age group is summarised in table 2.

{kind=link}
Graph showing percentage of patients in each age group with a particular diagnostic feature.
The number of affected subjects examined for disease features in each age cohort
PIGMENTARY FEATURES
Café au lait spots
Café au lait spots (CALS) were found in 434 out of 442 patients (98.2%), with 383 of these 434 (88.2%) possessing more than six. No information about CALS was available for 81 patients. One hundred and six out of the 120 NF1 patients (88.3%) in the 0-9 year age group had more than six spots, increasing to 110 out of 115 (95.7%) in the age group 10-19, and thereafter diminishing steadily. The younger patients fulfilled the NF1 diagnostic criteria by having other features of NF1 (Lisch nodules, plexiform tumour) and a family history. The older patients usually professed to having had CALS but that they had faded. Four patients fulfilled the criteria and so were included in the data but clearly had segmental disease (aged 12, 35, 52, and 65 years). One boy aged 12 years with four large CALS and no other features of NF1, but who had an affected parent and sib, was tested with intragenic markers. He had inherited the low risk haplotype from his affected parent. No other child aged 6 years or over with an affected parent has presented any difficulty in ascribing clinical status to them using the NIH criteria. Café au lait spots were usually the first sign to become apparent in young children and considered the most helpful in contributing to the diagnosis.
Axillary freckling
This was present in 310 out of 370 (87.8%) patients, with no information available for 153 patients. The proportion of affected adults (=>20 years) was 157 out of 175 (89.7%) and 153 out of 195 (78.5%) for affected children (<20 years).
Inguinal freckling
The proportion of patients possessing inguinal freckling was 151 out of 357 (42.3%), with no information available for 166 patients. Inguinal freckling was present in 80 out of 190 (42.1%) and in 71 out of 167 (42.5%) affected children and adults respectively.
Lisch nodules
Iris hamartomas were present in 157 out of 249 (63.1%) patients who had slit lamp examination. No information was available for 274 patients. The proportion of affected subjects appeared to increase with age but this may be owing to the small number of patients in the latter age groups, presenting an unstable estimate of the true proportion for these groups.
Neurofibromas
Cutaneous neurofibromas were reported in 217 out of 365 (59.4%) patients with 95 out of these 217 (43.8%) having 1-10 neurofibromas and the remaining 122 (56.2%) having more than 10. No information was available for 158 patients. The frequency with which cutaneous neurofibromas were reported increased throughout childhood and levelled off following early adulthood. The number of affected children was 62 out of 179 (34.6%) compared to 155 out of 186 (83.3%) affected adults.
Subcutaneous neurofibromas were reported in 150 out of 330 (45.5%) patients with 81 out of these 150 (54.0%) having 1-10 neurofibromas and the remaining 69 (46.0%) having more than 10. No information was available for 193 patients. These particular neurofibromas also seemed more prominent later on in life, occurring in 44 out of 170 (25.9%) affected children compared to 106 out of 160 (66.3%) adults. When neurofibromas were present in parous women they had been first noticed during pregnancy in four instances and were reported as having increased in size or number during that time in 10 cases, although this information was not specifically sought.
Plexiform neurofibroma
Eighty (15%) had one or more plexiform neurofibroma (nine patients had two, two had three, and one patient had more than three). A plexiform neurofibroma was present on the patient’s head or neck in 37% (24) of the 65 cases, 11% overall. Plexiform neurofibromas were found in 18% (42) of adults.
HEAD CIRCUMFERENCE
The median adult female head circumference was 56 cm (interquartile range=2 cm, range 53-62 cm, n=88) with 97% of the group equal to or below 61.33 cm. The median adult male head circumference was 59 cm (interquartile range=3 cm, range 53-64 cm, n=55) with 97% of the group equal to or below 63.32 cm. There was no information available for 22 adults.
THE EFFECT OF NF1 ON EDUCATIONAL PERFORMANCE
Of the 300 subjects for whom educational data were available, 18% (54) were attending or had required special school education. This included seven who were blind or partially sighted secondary to the effect of optic gliomas. Of the remaining cases, 34% (102) had received or were receiving normal school education but had specific learning difficulties and 30 needed extra help at school. Table 3 shows the results from an earlier study looking at a cohort of 417 of these patients where a variety of educational problems were identified. Problems with reading, writing, spelling, or mathematics were reported for 29% (60) of those for whom information was available or 14% of the total patient population.
Frequency of educational problems reported in a survey of genetic clinic and hospital notes from 417 patients carried out in NW England. For the purpose of this table each condition is taken to be exclusive
An educational difficulty or speech problem had been noted in 62% (300) of patients from whom information was available. If extrapolated to the whole NF1 population, 35% of all NF1 patients might be expected to experience some learning difficulties.
COMPLICATIONS
For the purpose of this study any condition which had previously been associated with NF1 in a large population or hospital based survey was considered to be a complication. The frequencies of major non-tumour complications associated with NF1 which were reported as either having occurred or being present in affected subjects in our study are shown in table 4. Other, less common complications mentioned in patient notes but not listed in table 4 were the following: buccal neurofibromas (three cases), parotid neurofibromas (three cases), pharyngeal neurofibromas (three cases), and sphenoid wing dysplasia (three cases). As sphenoid wing dysplasia is seldom sought as a confirmation of the diagnosis, it was not possible to determine a figure for how many patients were checked for this feature. Minor skin lesions such as capillary haemangioma were present in nine patients and vitiligo in three. The presence of hyperextensible or loose joints was reported in 15 subjects.
Frequency of non-tumour complications occurring in 523 patients with NF1 in a review of patient genetic clinic and hospital notes carried out in NW England
TUMOURS IN NF1
The tumours reported in the patients are shown in table 5. Optic gliomas were the most common CNS tumours. Of the malignant tumours not already listed in table 5 there was one case each of chronic myeloid leukaemia and rhabdomyosarcoma and two cases of breast cancer. Fifteen subjects had peripheral nerve malignancies of which the majority (8/15) were deep seated (thoracic, pelvic, pharyngeal). A total of 49 (9.4%) patients had malignant tumours related to NF1, six subjects having more than one. As an added check, the notes of all patients on the Regional Children’s Tumour (1956-1996) and Cancer Registries (1980-1996) have been reviewed.
Frequency of tumour complications occurring in 523 patients with NF1 in a review of patient genetic clinic and hospital notes carried out in NW England
ACTUARIAL ANALYSES
Optic glioma
In total, 25 of the 450 patients for whom complete follow up data to March 1996 were available had optic nerve glioma. Only 21 of the cases were included in the analysis as four patients had no date of diagnosis recorded. Table 6 shows the percentage of patients who were free from optic nerve glioma at the various time intervals shown.
Actuarial analysis showing percentage of 450 NF1 patients free from optic nerve glioma for the time intervals shown
Malignant nerve sheath tumours
In total, 15 of the 450 NF1 patients on whom we had follow up data were reported as having a malignant nerve sheath tumour. Table 7 shows the percentage of NF1 patients who were free from malignant nerve sheath tumours at the various time intervals shown. A further six cases of NF1 with optic glioma and six cases with MNST have been identified from the Children’s Tumour Registry and Cancer Registry. These cases have not been added to the actuarial analysis as they would have introduced selection bias. However, the absence of any other registrations in our NF1 population could be taken as further confirmation of tumour free status.
Actuarial analysis showing percentage of 450 NF1 patients free from a malignant nerve sheath tumour at the time intervals shown
Discussion
We have reported the findings in 523 subjects affected with NF1 in the north west of England. While ascertainment is not yet complete, we would expect a prevalence of around 1 in 4000-5000.10Referrals were largely from non-specialists and therefore this represents a relatively unbiased population. Population based studies are essential to arrive at an unbiased estimate of complications occurring in any disease. As the course of NF1 is unpredictable, even within a family carrying the same mutation, this study adds further data to enable accurate counselling of families. Families with NF1 are still being referred to our Genetic Register and so, although we have probably only ascertained around 70% for the expected prevalence, we feel that the study avoids the inherent bias of many hospital or specialist based clinic populations or where data are based on patients referred from a mixed population of specialist referral clinics.17 This is highlighted by the second analysis of our data when further numbers of patients were added. For most complications, the addition of up to 100 extra patients did not significantly alter the percentages, suggesting that the findings reflect true disease complication rates rather than bias in ascertainment.
FAMILY HISTORY
We were able to establish that 30.4% of patients with NF1 in our study had parents with no clinical features of the disease. It is our practice to examine parents’ skin completely and undertake slit lamp examination. This figure is comparable to the 30% reported in earlier population based studies.8 This is somewhat less than the one to one ratio one would expect from the high mutation rate,1 but may reflect that all referred subjects have a comprehensive family study performed in which mildly affected cases of NF1 maybe identified. Nonetheless, mildly affected subjects without a family history of NF1 are unlikely to present to the medical services and may therefore be under-represented in this and other similar studies. We are uncertain as to why more affected children are apparently born to affected mothers although the increased number was not significant at the 5% level (two tailed, p=0.14). Nevertheless, this observation has been reported by others8 and may reflect an under-reporting of families with an affected male parent or reduced genetic fitness in males.
One of the patients reported in this study had segmental NF1 and produced a daughter with unequivocal NF1. Other cases where a parent with segmental NF1 had produced affected offspring have been reported including the one in this study.18 19 It seems likely that gonadosomal mosaicism is the explanation for this observation.
DIAGNOSTIC FEATURES IN NF1
CALS, axillary and inguinal freckling, and Lisch nodules are usually the earliest signs to appear in a person carrying a defective NF1 gene. Results reported in this study for these diagnostic features are broadly similar to previous reports1 10 and axillary freckling was also more common than groin freckling. The importance of Lisch nodules as a diagnostic criterion for NF1 was only recognised just over a decade ago.20 21 Lisch nodules have been reported in other studies of NF118 22-23 where they were found to occur in approximately 75% of affected patients (although higher figures have also been reported). This was confirmed by our study. We have found no difficulty in ascribing clinical status using the NIH criteria and would concur with previous reports that penetrance is near complete by 5 years of age.8 9
Plexiform neurofibromas were present in 15% of patients in our study, a figure lower than that reported by Borberg24 and Croweet al 25 and substantially lower than the 53% occurrence reported by Riccardi.1 This difference might be explained by the fact that Riccardi1examined a selected patient population which had been referred to a specialist clinic and were thus more likely to have severe manifestations of NF1. A large number of the patients seen in this study were diagnosed only after a proband presented with NF1 and so often have fewer serious manifestations of the condition.
Cutaneous and subcutaneous neurofibromas were present in the majority of affected patients as found previously.16
HEAD CIRCUMFERENCE
Macrocephaly, which is a common feature in NF1,1 12 16 has been reported to have no adverse effect per se.1 These results confirm reports by other authors as to the frequency of macrocephaly in NF1.
EDUCATIONAL EFFECTS OF NF1
As with other studies which provide data on educational performance,1 8 12 26 we have relied upon evidence from patients and relatives on the educational problems encountered by patients with NF1. There seems little doubt that certain problems arise more frequently than can be accounted for by chance, for example, difficulties with language development. The type of educational assistance received by children with learning difficulties will vary according to their school or Local Education Authority’s policy, together with the availability of teachers to provide remedial education. Consequently any direct comparison with previously reported data which categorise children by the type of educational assistance that they receive would seem inappropriate. If more accurate data are to be obtained, long term prospective studies will need to be undertaken using assessments by educational psychologists, speech therapists, geneticists, and other clinical staff. However, more comprehensive studies of psychometric analysis are becoming available.27 28
COMPLICATIONS
The frequency with which serious complications occur in childhood varies between those which are present in 1-6% of patients, such as pseudoarthrosis requiring surgery (1.9%) to severe plexiform neurofibromas on the head and neck (6.0%), and those which are rare and occur in less than 1%. The results for all serious complications are similar to those reported in other studies.8 24
Treatable complications which can occur at any age and were noted in more than 1% of the study group were: epilepsy, spinal neurofibromas, aqueductal stenosis, and visceral neurofibromas, all of which have been reported by others with similar frequencies.8
TUMOURS
Both types of neurofibromas, subcutaneous and cutaneous, are said to increase in number with age between early and mid adulthood.1 They may undergo malignant change26 27 although this is more likely in the larger plexiform neurofibromas.29 30 It was not possible to verify that the MNSTs seen in the study occurred in pre-existing plexiform neurofibromas as the data were taken from notes and pathology reports and so the necessary information was not always present. In the actuarial analysis, it is likely that the data shown are an accurate reflection of MNST in NF1 until the age of 50 years. While there may be a bias in referral in those patients and families where a major complication has been described, there is also likely to be some underascertainment in patients in whom the diagnosis of NF1 is made at the time their MNST is diagnosed. The poor prognosis associated with the tumour means that some of these people will not survive to be referred. Beyond the age of 50 years, the numbers are small and, combined with possible ascertainment bias for those with problems in this age group, these figures are probably a less accurate representation of true incidence. Nevertheless, even taking some degree of bias into account, these results imply a much higher overall risk of peripheral MNST than many previous studies (cross sectional studies will always underestimate the risk of a tumour with poor survival). It is important that any information provided to patients about the risks of serious complications, such as malignant change to an MNST, should be as accurate as possible to prevent unnecessary anxiety. The actuarial figure calculated for the risk of developing an MNST here is high. To try to validate the risk figure further, an analysis of patients with NF1 and MNST in the north west region has been undertaken. Using information from the North West Tumour Registry and the NF1 Genetic Register, a total of 13 MNST were identifed in patients with NF1 over a 10 year period. The population in this region is 4 million, and assuming a frequency of NF1 in the population of 1 in 3000, there would be 1330 cases of NF1. This is approximately equivalent to a 1 per 1000 annual incidence rate of MNST. During a lifetime of 75 years, the overall risk of an MNST in a patient with NF1 would be 7.5%. This figure assumes complete ascertainment of all NF1 related MNSTs, which is unlikely. It also assumes a maximum frequency for NF1 in the population. Overall, while the actuarial risk calculated is likely to be an overestimate, taking these population data into account, the true risk figure probably lies between 7.5% and 15%.
This means that any suspicious change in neurofibromas warrants careful examination, with biopsy if necessary. Nonetheless the tumours are often deep seated and the diagnosis is still delayed, as it was in at least 7/15 of our cases. Patients at the clinic are asked to present quickly if an existing neurofibroma enlarges or becomes painful. Despite an often early presentation, the patients who were diagnosed prospectively with an MNST did not have a better outlook than those who presented without previous knowledge of the possible malignant change. This reflects the aggressive nature of the tumour.
Actuarial risks were also calculated for optic nerve gliomas. There were some limitations on the data with which to calculate these figures. To ensure complete ascertainment and follow up and so statistically validate the data, some patients were excluded. This was mainly attributed to failure to attend follow up appointments or return regular update letters, lost notes, notes out to review and not available, or incomplete documentation in the patient’s notes. Treatment of a first tumour in a condition which predisposes to tumours may also increase the risk of a second tumour. No evidence for this was seen, mainly as those patients who had aggressive treatment had poor survival. It is, nevertheless, an important consideration.
In this study CNS tumours occurred in 9.4% (49) of patients, with optic gliomas constituting the majority of these. All these gliomas were either symptomatic or were detectable by abnormalities on ophthalmological examination. Analysis of the Regional Children’s Tumour Registry has identified a further six cases of optic glioma said to be associated with neurofibromatosis type 1. These cases have not been included in these data as they were not referred to the register and have not been seen in our clinics. Although previous reports for the incidence of optic gliomas have varied between 0.7%10and 15%,1 31 32 these latter studies screened asymptomatic subjects with CT. However, follow up studies with the one series of 33 patients with optic gliomas on scan33 has shown progression in only three of these tumours over a mean period of 2.4 years. These all occurred in the symptomatic group who were all below 6 years of age. The tumours which were symptomatic and needed treatment in this study were similarly under the age of 6 years. Those diagnosed in the older group were usually found on cerebral imaging for other indications. It would therefore seem likely that the majority of asymptomatic tumours never cause problems and almost certainly regress in adulthood.33 Regression in adult life without treatment has been previously reported.34 It is also advisable with the increasing availability of MRI scanning that this should be used in place of CT to avoid the possible risks of radiation induced tumours, particularly in asymptomatic subjects. There are differences of opinion as to whether symptomatic cases benefit from treatment,35although radiotherapy and chemotherapy have been shown to be beneficial in tumour management.36 The diagnosis of an asymptomatic tumour which may never affect the patient will only serve to increase the level of anxiety in both patient and parents alike. It would therefore seem more appropriate to operate a policy of careful clinical surveillance rather than scannng all children with NF1.33
Vestibular schwannoma did not occur in any patient in our study. This supports evidence from recent population based studies that this is not a feature of NF1.16 Previous studies which have quoted an incidence for this tumour were contaminated with neurofibromatosis type 2 (NF2) cases,25 which is a genetically and phenotypically distinct condition. Other features of NF2, such as schwannomas of cranial, spinal, and peripheral nerves as well as cranial and spinal meningiomas (only one case of intracranial meningioma at population risk level), were also absent confirming other recent studies16 and emphasising the fundamental difference between NF1 and NF2.
Conclusions
The absence of any clear genotype/phenotype correlation in NF1 and the lack of information on modifying genes means that population based clinical studies will continue to provide an important source of information for patient and family management and research.
Acknowledgments
We would like to thank our colleagues at the CRC Paediatric and Familial Cancer Research Group at the Royal Manchester Children’s Hospital for their help with the setting up of the database and in obtaining the necessary data. We would also like to thank our clinical colleagues involved in the running of the NF1 Genetic Family Register Service.