Article Text

Abstract
INTRODUCTION Congenital disorders of glycosylation (CDG), or carbohydrate deficient glycoprotein syndromes, form a new group of multisystem disorders characterised by defective glycoprotein biosynthesis, ascribed to various biochemical mechanisms.
METHODS We report the clinical, biological, and molecular analysis of 26 CDG I patients, including 20 CDG Ia, two CDG Ib, one CDG Ic, and three CDG Ix, detected by western blotting and isoelectric focusing of serum transferrin.
RESULTS Based on the clinical features, CDG Ia could be split into two subtypes: a neurological form with psychomotor retardation, strabismus, cerebellar hypoplasia, and retinitis pigmentosa (n=11), and a multivisceral form with neurological and extraneurological manifestations including liver, cardiac, renal, or gastrointestinal involvement (n=9). Interestingly, dysmorphic features, inverted nipples, cerebellar hypoplasia, and abnormal subcutaneous fat distribution were not consistently observed in CDG Ia. By contrast, the two CDG Ib patients had severe liver disease, enteropathy, and hyperinsulinaemic hypoglycaemia but no neurological involvement. Finally, the CDG Ic patient and one of the CDG Ix patients had psychomotor retardation and seizures. The other CDG Ix patients had severe proximal tubulopathy, bilateral cataract, and white matter abnormalities (one patient), or multiorgan failure and multiple birth defects (one patient).
CONCLUSIONS Owing to the remarkable clinical variability of CDG, this novel disease probably remains largely underdiagnosed. The successful treatment of CDG Ib patients with oral mannose emphasises the paramount importance of early diagnosis of PMI deficiency.
- CDG
- phosphomannomutase
- phosphomannose isomerase
- dolichyl-phosphate glucose:mannose 9 N-acetylglycosamine 2 glucosyltransferase
Statistics from Altmetric.com
- CDG
- phosphomannomutase
- phosphomannose isomerase
- dolichyl-phosphate glucose:mannose 9 N-acetylglycosamine 2 glucosyltransferase
Congenital disorders of glycosylation (CDG), or carbohydrate deficient glycoprotein syndromes, form a recently recognised group of inherited multisystem disorders characterised by the abnormal glycosylation of a number of serum glycoproteins and comprising two types.1 ,2 In CDG I, the most frequent form of the disease,3 ,4 serum transferrin is composed of normally glycosylated and partially or totally hypoglycosylated molecules.2 In 70% of CDG I patients (CDG Ia), this structural abnormality is related to deficient phosphomannomutase (PMM) activity,4 ,5 and mutations of thePMM2 gene have recently been identified.6 ,7 However, phosphomannose isomerase deficiency (PMI) has been found in a fraction of CDG I patients harbouring normal PMM activity (CDG Ib)8 ,9 while a dolichyl-phosphate glucose:mannose 9 N-acetylglycosamine 2 glucosyltransferase deficiency, caused by mutations in the human orthologue of yeast ALG6 (CDG Ic),10 ,11 a DolPP-Man5GlcNAc2 dependent mannosyltransferase deficiency, caused by mutations in the geneALG3 (CDG Id),12 and a dolichylmannose synthase-1 deficiency, caused by mutations in theDPM1 gene (CDG Ie)13 ,14 were found in other type I patients. CDG II differs from CDG type I in the glycosylation pattern of serum transferrin and is the result of defects in the processing of protein bound glycans.15-17
Here, we report the clinical and biological features of 26 CDG I patients, including 20 patients with CDG Ia, two with CDG Ib, one with CDG Ic, and three with unclassified CDG Ix, and we provide additional support for the clinical variability of the disease. CDG Ia causes neurological disorders, including psychomotor retardation, cerebellar hypoplasia, and strabismus with or without multivisceral involvement.1 ,18 By contrast, CDG Ib causes enterohepatic disorders with no neurological involvement, and this is the only form responding to oral mannose administration.8 ,9 ,19 The other types are rare and present as neurological diseases.10-12
Methods
Twenty six patients from 22 unrelated families were examined in either our hospital (n=20) or other French paediatric units (n=6). Criteria for inclusion were an abnormal CDG I glycosylation pattern of serum glycoproteins, identified by western blotting assay20 of serum transferrin, haptoglobin, α1-acid glycoprotein, and α1-antitrypsin on filter paper blood spots, or isoelectric focusing2 ,21 of serum transferrin or both.
Cytosolic PMM and PMI activities were determined at 37°C according to van Schaftingen and Jaeken4 in either blood mononucleated leucocytes or cultured skin fibroblasts and dolichyl-phosphate glucose:mannose 9 N-acetylglycosamine 2 glucosyltransferase activity was determined according to Korner et al.10 Mutations in thePMM2, PMI, andALG6 genes were identified by sequencing the specific cDNA after extraction of total mRNA from cultured skin fibroblasts or circulating lymphocytes.7 ,11 ,19
Results
CLINICAL FINDINGS
A total of 26 patients (14 females and 12 males) ranging in age from 3.5 months to 19 years were included in the study. Their clinical features are summarised in tables 1 and 2. Based on both the glycosylation pattern of serum N-glycoproteins and the results of enzymatic and molecular studies, the 26 CDG patients were classified into different subtypes (tables 1 and 2), namely PMM deficiency (CDG Ia, patients 1-20), PMI deficiency (CDG Ib, patients 21-22), dolichyl-phosphate glucose:mannose 9 N-acetylglycosamine 2 glucosyltransferase deficiency (CDG Ic, patient 23), and non-a, non-b, non-c CDG I (CDG Ix, patients 24-26). Patients 3 and 4 were sibs, as were patients 10 and 11, 16 and 17, and 18 and 19. Patient 8 had two affected sisters, who were not included in the study. Among the families living in France, three were of Portuguese ancestry (patients 3, 4, 16, 17, 18, and 19) and one was of Lebanese origin (patient 21).
Clinical manifestations and laboratory investigations in 20 CDG Ia patients
Clinical manifestations and laboratory investigations in CDG Ib, Ic, and Ix patients
CDG IA (PATIENTS 1-20)
Two presentations of the disease were recognised among CDG Ia patients: a neurological form (1-11) and a multivisceral form including neurological and extraneurological manifestations (12-20, table 1). Parameters at birth were normal in all but two cases (18 and 19).
In the patients with the neurological form (1-11), the first clinical manifestations occurred within the first year of life and included neonatal hypotonia (8/11), strabismus (10/11), ataxia (11/11), and psychomotor retardation (11/11). However, hypotonia and strabismus were not consistently present or were ignored until the patients were referred to a paediatric unit because of psychomotor retardation and cerebellar hypoplasia at 6 months of age. Age at diagnosis was relatively advanced in most cases (table 1). Areflexia (9/11) and retinopathy (7/11) were frequent features after 1 year of age, while non-neurological symptoms were occasionally observed (hepatic, cardiac, renal, or gastrointestinal, table 1). Patients 10 and 11 also had nystagmus. Severe psychomotor retardation was present in all the patients but one (patient 6). The oldest patients (3, 4, 5, 8, 10, and 11) attended special schools and could walk unaided aged 2 and 7 years, but patients 10 and 11 could not walk aged 6-9 years. Facial dysmorphic features (prominent forehead, large ears, thin upper lip), inverted nipples, and abnormal adipose tissue distribution with combined lipohyperplastic and atrophic changes were observed in the youngest patients only (patients 1, 3, 4, 5, figs 1 and 2). In the oldest patients, kyphosis and limb joint restriction were also observed, along with absence of puberty and lack of secondary sexual characteristics related to primary ovarian failure in the two oldest girls (4 and 5, fig 3). All the patients are still alive.

Facial features of eight patients with CDG Ia before 2 years of age. They all share common features including a high forehead, a very thin upper lip, and large and floppy ears. Note that patients 12 and 13 have upward slanting palpebral fissures while patients 2 and 15 have downward slanting palpebral fissures. (A) Patient 12. (B) Patient 13. (C) Patient 14. (D) Patient 15. (E) Patient 1. (F) Patient 4. (G) Patient 2. (H) Patient 3.

Classical features of CDG type I including inverted nipples (A) and abnormal fat distribution (C, D) with fat pads (B).
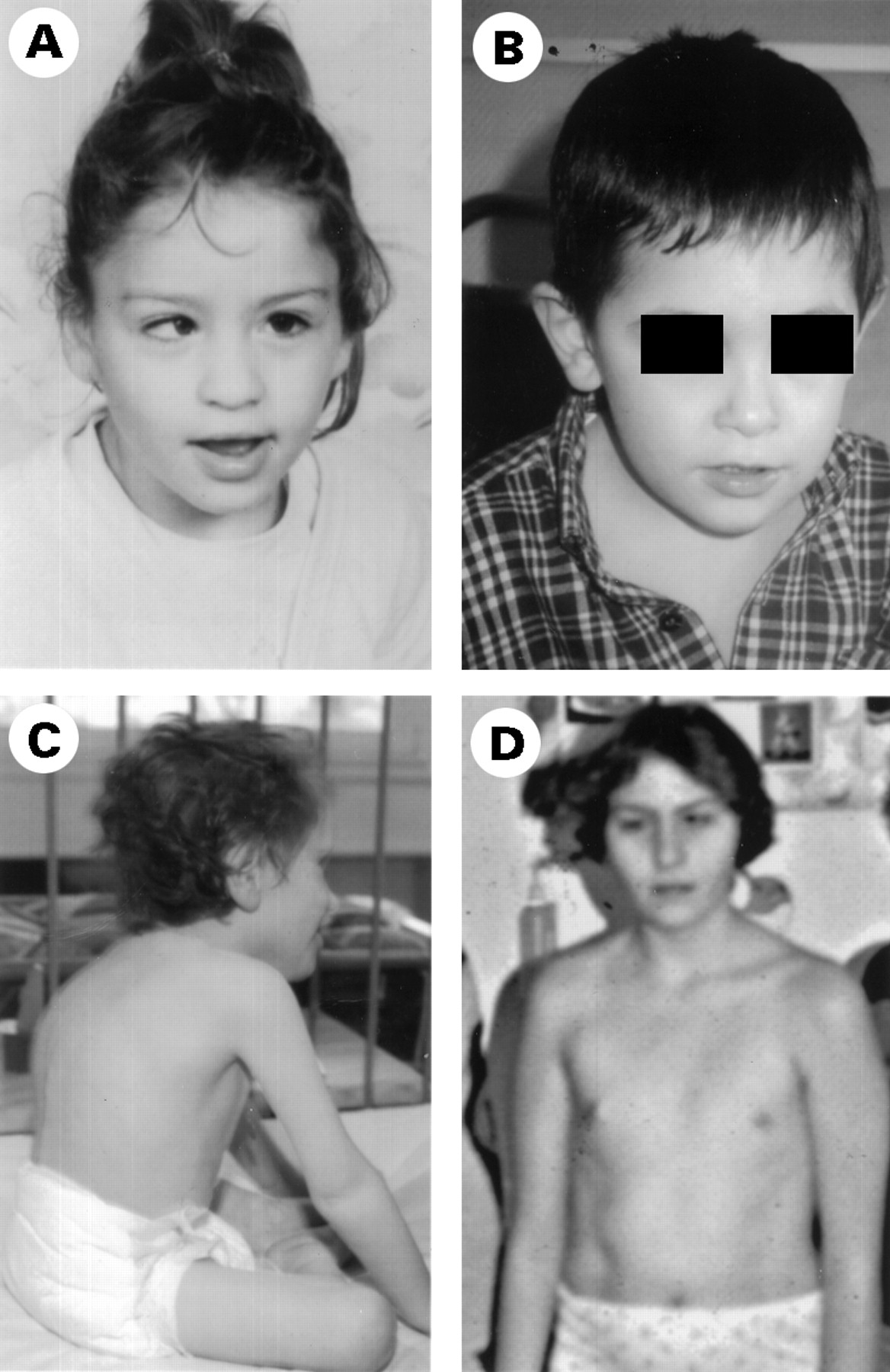
Evolution of the facial features. The same girl (patient 4) is shown in fig 1F at the age of 18 months and in fig 3 at the age of 6 years (A) and 16 years (D). The only notable features are strabismus and a thin upper lip at 16, and absence of breast development owing to hypogonadism. In patient 6 (B), note thin upper lip but no other facial dysmorphic features at 5 years. In patient 15 at 6 years (C), note the kyphosis and flexion deformity of the knee.
In the neurological-multivisceral form (patients 12-20), the patients were referred to a paediatric centre for failure to thrive with diarrhoea (4/9), pericarditis (2/9), liver disease (2/9), or proximal tubulopathy (1/9, table 1). Onset occurred before the age of 3 months, but the diagnosis was made later in life or even after death (16, 18, 19). The two sibs who displayed liver involvement (18, 19) developed hepatomegaly, vomiting, and liver damage during their first days of life and died of liver failure. Two sibs and an unrelated child had pericarditis at one week (16, 17) and 18 months respectively (12) and died before 2 years of age of their heart disease. The patients who survived (5/10) developed failure to thrive, hepatomegaly, and psychomotor retardation similar to the neurological form in the course of their disease. Patient 14 had severe enteropathy with diarrhoea, steatorrhoea, and recurrent episodes of fever related to low serum immunoglobulin levels requiring immunoglobulin infusions. Patient 20 had severe proximal tubulopathy but renal hyperechogenicity was present in all patients and histopathological analysis of the liver detected fibrosis in all patients tested (steatosis in 3/4 cases, cirrhosis in 1/4 case). By contrast, intestinal villous atrophy was found in only 2/6 patients tested (18-19). Finally, cerebellar hypoplasia, strabismus, facial dysmorphism, inverted nipples, and skin defects were frequent (fig 1 and 2), but were not present in two patients (patients 18 and 19).
CDG IB (PATIENTS 21 AND 22)
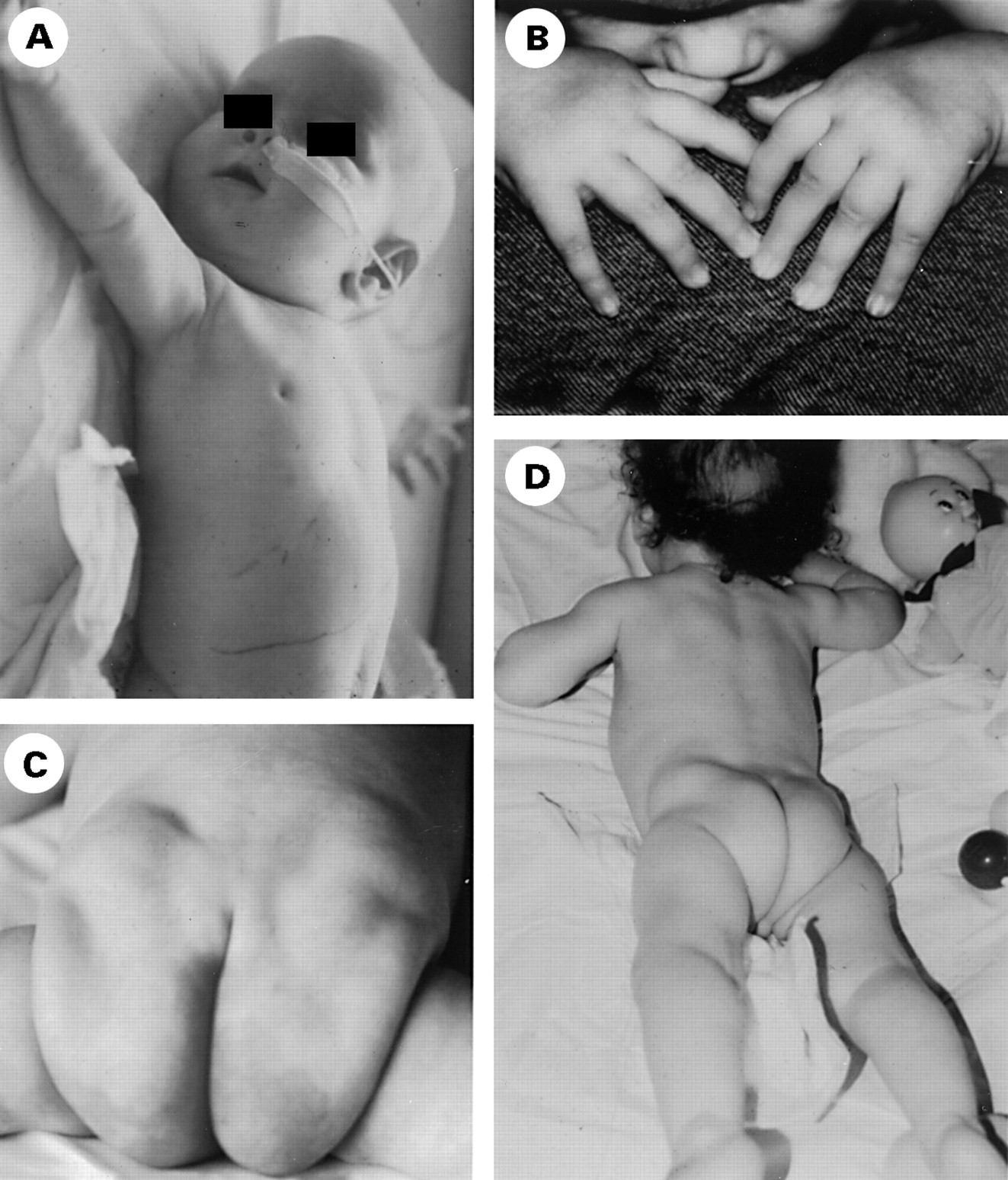
The two CDG Ib patients were referred for enteropathy and hypoglycaemia respectively (table 2). Both had liver (hepatomegaly) and gastrointestinal involvement (vomiting and diarrhoea) and hyperinsulinaemic hypoglycaemia at 3 months of age. One patient also had right auriculothrombosis and required large amounts of glucose (12 mg/kg/min, patient 22) to maintain adequate blood glucose levels.22 Oral mannose administration significantly improved his clinical status.22 ,23 The other one had recurrent cholangitis and decreased immunoglobulin levels (patient 21) and died at 3 years of age of liver failure. Histopathological studies showed liver fibrosis, steatosis, and cirrhosis, with villous atrophy and lymphangiectasia. No neurological, cutaneous, or dysmorphic features were noted (fig 4A, B), but both patients had renal hyperechogenicity.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Facial features of CDG Ib and Ix. Note high forehead in CDG Ib patients 21 (A) and 22 (B). (C) Patient 24 (Ix). Note high forehead, very thin upper lip, and bitemporal retraction. (D) Patient 26 (Ix). Note nystagmus and coarse face.
CDG IC AND IX (PATIENTS 23-26)
One patient with CDG Ic (patient 23) and three CDG Ix patients (patients 24-26) suffered psychomotor retardation. Two of them also had seizures (patients 23 and 24, table 2). Their presenting manifestations included retinitis pigmentosa and psychomotor retardation at 8 months of age and they are still alive at 2 and 10 years of age, respectively. Patient 24 still cannot walk (fig 4C), while patient 23 started to walk aged 7 years. Another patient (patient 25) presented with mild psychomotor retardation associated with severe proximal tubulopathy at 2 months of age, bilateral cataract, white matter abnormalities, and renal hyperechogenicity. The last patient (patient 26, fig 4D) was referred for oedema related to hypoproteinaemia at 1 month of age. She had neonatal hypoglycaemia and thrombocytopenia, severe encephalopathy, nystagmus, coarse face, diarrhoea, mild septal hypertrophy, iris coloboma, bilateral cataract, liver fibrosis, renal hyperechogenicity, and micropolygyria (table 2). She developed West syndrome and died at 6 months of age.
BIOCHEMICAL AND MOLECULAR INVESTIGATIONS
Serum electrolyte and creatinine levels were normal but liver and clotting functions were altered in CDG patients. Serum transaminase activities were increased in 23/26 patients (AST and ALT 1.5 × normal) and increased during episodes of fever. Hypoalbuminaemia (<30 g/l) related to liver dysfunction was present in 9/26 patients and was responsible for oedema in four of them. Hypocholesterolaemia (mean 2.2 mmol/l, normal >3.2 mmol/l) was found in 3/10 type Ia patients and clotting tests showed thrombocytosis in half of them (>600 000 platelets/mm3).
Serum glycoproteins including coagulation factor XI (<50%, normal 70-130) were consistently decreased and factor XII was also low in one patient (32%, normal 70-130), resulting in a raised prothrombin time (>10 seconds). Serum immunoglobulins A and G were reduced in half of the patients (A: 0.05 to 0.20 g/l, normal >0.4 g/l; G: mean 2.5 g/l, normal >4 g/l). Hypothyroidism in 3/8 CDG Ia patients was ascribed to a combination of low thyroxine binding globulin (mean 7 mg/l, normal range 10-40 mg/l), with slightly increased basal thyroid stimulating hormone (6 to 10 mU/l, normal range 0.5-3 mU/l) and reduced thyroxine levels (8-12 pmol/l, normal range 10-20 pmol/l). Interestingly, no hypothyroidism was found in type Ib patients. The two oldest female patients (4 and 5) had hypergonadotrophic hypogonadism and no detectable ovaries. Low oestradiol concentrations (<10 pg/ml) were associated with high levels of serum luteinising hormone (15 μU/ml) and follicle stimulating hormone (48 μU/ml) concentrations.
Tables 1 and 2 summarise the results of thePMM2 and PMImutation screening in our series. The R141H missense mutation was detected in the PMM2 gene of 17/19 CDG Ia patients (table 1) and all of them were compound heterozygotes. The I132T mutation was observed in three unrelated patients with the neurological form while the V231M mutation was present in two unrelated families with the multivisceral form of the disease.
Discussion
We report the clinical and biological features of 26 CDG patients (20 Ia, two Ib, one Ic, and three Ix) born to 22 unrelated families living in France. CDG Ia, the first described and the most frequent form of the disease,1 was associated with two clinical presentations: a solely neurological form and a neurological-multivisceral form.18 ,24 In the neurological form, strabismus, psychomotor retardation, and cerebellar hypoplasia occurred early, while neuropathy and retinitis pigmentosa were frequently observed in the course of the second year of life. Surprisingly, this form was not fatal while in the multivisceral form, neurological and extraneurological manifestations occurred early in life and were frequently fatal. All organs but the lungs could be involved, explaining why several patients were primarily referred to specialised units (for example, pericarditis, severe hepatocellular failure, chronic diarrhoea, or proximal tubulopathy).24-26 Hepatic fibrosis and renal hyperechogenicity were consistent features while strabismus and cerebellar hypoplasia were occasionally absent. Relatively specific symptoms of CDG Ia, including dysmorphic features, inverted nipples, and abnormal fat pads, were occasionally absent in both forms.24-26 However, the clinical presentations were largely homogeneous within CDG families.
The heterozygous R141H PMM2 mutation was present in most CDG Ia patients of French and Portuguese ancestry, regardless of their clinical presentation (neurological or multivisceral).27 No patient was homozygous for this mutation suggesting that the R141H mutation is a severe mutation and that homozygosity may not be compatible with life.27 The I132T PMM2 mutation was found in three unrelated patients with the neurological form, while the R141H/V231M genotype was observed in two families with the multivisceral form of the disease. The other 17 mutations were scattered throughout the gene, hampering genotype-phenotype correlation.27
While both CDG Ia and Ib patients had an altered serum transferrin glycosylation pattern with decreased serum coagulation glycoprotein concentrations, liver abnormalities, and renal hyperechogenicity, their clinical manifestations were strikingly different, as previously reported.8 ,9 ,22 ,23 In CDG Ib, the main clinical features included liver disease, enteropathy, and hyperinsulinaemic hypoglycaemia, while neurological symptoms and lipocutaneous abnormalities were absent. Thrombosis has also been described.8 In contrast with CDG Ib, psychomotor retardation with no cerebellar hypoplasia was the main feature in CDG Ic and CDG Ix.
The successful treatment of patients with CDG Ib with oral mannose emphasises the primary importance of early diagnosis of PMI deficiency. Mannose 6-phosphate, the precursor of mannose1-phosphate and GDP-mannose (the mannose donor for glycoprotein glycan synthesis), can stem from either fructose 6-phosphate (via PMI and the glucose pathway) or mannose phosphorylation (via hexokinase). Mannose intake produces mannose 6-phosphate directly and therefore bypasses the PMI deficiency.28-33 However, mannose overload can be toxic for the kidneys and the long term effects of oral mannose are unknown.
In conclusion, the various forms of CDG are remarkably different in their clinical course and outcome. CDG Ia accounts for two main forms, namely a neurological and a multivisceral form, with markedly different prognoses. The broad spectrum of CDG manifestations suggests that this condition is largely underdiagnosed. This diagnosis should not only be considered in cerebellar hypoplasia with abnormal subcutaneous fat distribution and inverted nipples, but also when dealing with psychomotor retardation, retinitis pigmentosa, seizures, and multiorgan failure. Liver and clotting abnormalities should help in diagnosing this condition18 ,24 and it should be screened for by western blotting assay of glycoproteins or isoelectric focusing of transferrin on filter paper blood spots or serum. Measurement of enzymatic activities and molecular studies will then definitely confirm the diagnosis.