Article Text

Abstract
Objective To document the cardiac phenotype associated with Friedreich’s ataxia, a recessively inherited disorder characterised by spinocerebellar degeneration.
Setting Individuals with Friedreich’s ataxia who accepted the invitation to participate in the study.
Hypothesis The cardiomyopathy associated with Friedreich’s ataxia may offer a human model for the study of factors modulating cardiac hypertrophy.
Methods 55 patients (mean (SD) age 30 (9) years) with a clinical diagnosis of Friedreich’s ataxia were studied by clinical examination, electrocardiography, cross sectional and Doppler echocardiography, and analysis of the GAA repeat in the first intron of the frataxin gene.
Results A wide variety of cardiac morphology was documented. Subjects with normal frataxin alleles had no evidence of cardiomyopathy. In homozygous subjects, a relation was found between the thickness of the interventricular septum (r = 0.53, p < 0.005), left ventricular mass (r = 0.48, p < 0.01), and the number of GAA repeats on the smaller allele of the frataxin gene. No relation was shown between the presence of electrocardiographic abnormalities (mainly repolarisation changes) and either the pattern of ventricular hypertrophy (if present) and degree of neurological disability or the length of time since diagnosis. No tendency to ventricular thinning or dilatation with age was found. Although ventricular systolic function appeared impaired in some cases, Doppler studies of ventricular filling were within the normal range for age.
Conclusions The cardiomyopathy associated with Friedreich’s ataxia shows a variable phenotype which is not concordant with the presence of ECG abnormalities or the neurological features of the condition. As the genetic basis for Friedreich’s ataxia has been established, further studies will help to clarify the molecular mechanisms of the cardiac hypertrophy.
- cardiomyopathy
- Friedreich’s ataxia
- genetics
Statistics from Altmetric.com
Friedreich’s ataxia is the most prevalent of the spinocerebellar ataxias and is inherited as an autosomal recessive disorder. Cardiac abnormalities are generally accepted to be part of the condition,1-5 with studies of the cardiac phenotype reporting variable left ventricular hypertrophy (concentric, asymmetric, or both3 ,6-12) and left ventricular outflow tract obstruction.13 In some reports the cardiac abnormalities have been extreme enough to warrant the description of the “hypertrophic cardiomyopathy of Friedreich’s ataxia,”14 but such studies have been based on relatively small numbers of affected individuals. It is unclear why patients with Friedreich’s ataxia should develop gross cardiac hypertrophy. Familial hypertrophic cardiomyopathy is a dominantly inherited condition associated with mutations in a number of genes encoding sarcomeric proteins in the myocardium; there are no extracardiac features except skeletal muscle abnormalities.15-21 In contrast, Friedreich’s ataxia is a genetically homogeneous disorder in which degeneration of the posterior columns, the corticospinal and spinocerebellar tracts, and cardiac abnormalities occur together. A gene, X25, has recently been reported which encodes a 210 amino acid protein, frataxin. Expression of the gene appears to be highest in the primary sites of degeneration, and the clinical features of Friedreich’s ataxia reflect a reduction in frataxin synthesis associated with the expansion of a GAA trinucleotide repeat in the first intron of the gene in spinal cord, heart, and pancreas.22 Recent reports suggest that frataxin plays a central role in the regulation of mitochondrial iron transport.23 ,24
This prospective study was undertaken to characterise the cardiac phenotype and molecular genetic abnormalities in Friedreich’s ataxia, in order to elucidate the factors contributing to the development of myocardial hypertrophy in this disorder.
Methods
Patients with a clinical diagnosis of Friedreich’s ataxia were invited to attend for cardiovascular studies with their immediate relatives (particularly parents) where practicable. The study was undertaken over a three month period and data were obtained from 55 patients who satisfied the Harding clinical criteria for the diagnosis of Friedreich’s ataxia.25 ,26 All had onset of neurological symptoms before the age of 25 years (although one subject was included with onset at 26 years), progressive unremitting ataxia of gait of all limbs, and absent lower limb reflexes, dysarthria, and extensor plantar responses. The diagnosis had previously been confirmed by an experienced neurologist (the late professor A Harding) following nerve conduction studies in all but two cases. The mean (SD) age of onset of initial symptoms (usually ataxia on running) was 12 (5) years (range 3–26), with a mean duration of neurological symptoms of 17 (8) years at the time of the study. Thirty eight (70%) were confined to a wheelchair (20 were totally unable to stand), and 84% were heavily dependent on others for the activities of daily living.
Standard 12 lead ECGs were recorded in the recumbent position. The echocardiographic studies were all performed by one operator (JED) using an Acuson 128 XP10 scanner. The echocardiogram was recorded in a conventional manner with occasional modification of the transducer site on the chest wall to optimise image quality in four subjects with uncorrected kyphoscoliosis which precluded satisfactory imaging from the conventional parasternal window. Measurements were made from the M mode recordings according to the standards of the American Society of Echocardiography and analysis was performed using the software programs of the scanner.
Doppler studies were performed using pulsed ultrasound waveforms generated from either the 2.5 or the 3.5 MHz transducer. For studies of left ventricular filling, the sample volume was placed in the left ventricle approximately 1 cm above the mitral valve annulus and the position optimised to ensure that the maximum diastolic flow velocities were recorded. The left ventricular outflow tract (LVOT) studies were recorded with the sample volume just below the aortic valve. Peak aortic velocities were recorded using continuous wave Doppler across the aortic valve. The variables measured were the peak aortic and LVOT velocities, the velocity time integral, maximun acceleration, and time to peak velocity of the aortic, LVOT, and transmitral flow. The diastolic waveform of left ventricular filling was also used to calculate the early peak flow velocity (E), the peak late (atrial) diastolic flow velocity (A), the E/A ratio, the deceleration time of early diastolic filling, and the isovolumetric relaxation time. The measurements were made from four consecutive cardiac cycles and the results averaged. All the recordings were made by one operator using the online facility of the scanner.
ANALYSIS OF THE GAA REPEAT
Long range polymerisation chain reaction (PCR) techniques were employed to amplify the fragment of DNA incorporating the GAA triplet repeat. DNA was extracted from blood leucocytes and size of the GAA triplet repeat assessed using the amplifiers 5′-GGGATTGG TTGCCAGTGCTTAAAAGTTAG-3′ and 5′-GATCTAAGGACCATCATCATGGCCACA CTTGCC-3′ to generate a product of 457 base pairs plus the number of base pairs comprising the GAA expansion. The ELONGASE enzyme mix (Taq and pyrococcus species polymerases, GibcoBRL, Paisley, UK) was used for amplification (35 cycles; 94°C for 30 seconds, 60°C for 30 seconds, and 68°C for 120 seconds). The PCR products were separated by electrophoresis through a 1% agarose gel. The results were confirmed using the Expand high fidelity PCR system (Taq and Pwo polymerases, Boehringer Mannheim, Lewes, UK) using 10 cycles of 94°C for 15 seconds, 60°C for 30 seconds, and 68°C for 120 seconds, followed by 25 cycles where the 68°C step was lengthened by 20 seconds/cycle. The PCR products were transferred to Hybond N+ and the size of the GAA expansion confirmed by hybridisation with 100 ng of a (GAA)10 oligonucleotide end labelled with 1 μl of [γ32P] ATP (5000 Ci/mmol; Amersham, UK).
STATISTICAL METHODS
The results were analysed using Student’st tests with the Bonferroni correction to take account of multiple comparisons. Trends were assessed by linear rank correlation and the results are presented as mean (SD) unless otherwise indicated. The significance level, α, was set at 0.05.
Results
The demographic features of the study patients are given in tables1 and 2. Breathlessness on exertion was reported by 10 subjects, eight of whom were not confined to a wheelchair. Palpitations were reported in eight subjects and three had been prescribed a β blocker for intermittent use. In one individual a diagnosis of cardiomyopathy had been made by a physician with an interest in cardiology following echocardiography at the local district general hospital and treatment included an angiotensin converting enzyme (ACE) inhibitor. Clinical examination of the heart was normal in all except four patients, one with an ejection systolic murmur at the left sternal margin (and a peak velocity in the left ventricular outflow tract of 1.8 m/s but no ventricular hypertrophy), and three had a soft pansystolic murmur of mitral regurgitation. All were in sinus rhythm and none of the subjects was hypertensive (mean blood pressure 116 (12)/73 (5) mm Hg).
Demographic features of the 55 subjects (25 male, 30 female): entire group of ataxic subjects
Demographic features of the 55 subjects, classified by presence of GAA expansion
GAA EXPANSION
Forty five subjects were homozygous for the GAA expansion while in the 10 remaining there was no evidence of expansion in either allele of the frataxin gene (table 2). None of the subjects studied was heterozygous for the GAA expansion. The median number of triplet repeats in the smaller expanded allele was 700, with a range of 230 to 1200. There was no relation between the size of the expansion and age of onset or age when confined to a wheelchair, with similar ranges being found in those individuals without expansions.
ELECTROCARDIOGRAPHY
The ECG findings are summarised in table 3 and an example is given in fig 1. The ECG was normal in eight individuals who all had a normal allele; otherwise the most common abnormality was T wave inversion. Inferior changes were present in 44% and lateral changes in 47%, with widespread T wave inversion in 20 cases. In three cases the R wave in aVL exceeded 11 mm and the QRS axis was between 0° and −30°, but this was associated with echocardiographic left ventricular hypertrophy in only one of these cases.
Electrocardiographic findings in Friedreich’s ataxia

Twelve lead electrocardiogram from an 18 year old patient with Friedreich’s ataxia (800 and 1160 GAA repeats on the frataxin alleles), showing inferolateral T wave and deep S waves in the septal leads.
Eight subjects had a resting sinus tachycardia with a pulse rate of > 90 beats/min; two had a normal allele. There was no evidence of pre-excitation in any case, and right bundle branch block was present in one (with partial right bundle branch block in another case in addition). In the group as a whole there was wide variation in the electrical axis of the heart, the mean QRS vector being 76° (SD 54°; range −58° to +240°), with a predominance of right axis deviation (> 90° in 31%). There was also considerable variation in the P wave axis (−16° to +84°) and the T wave axis (−86° to +268°). The mean difference between the QRS and T wave axis was 72° (SD 65°; range 0° to 227°), the difference being > 60° in 22 cases. In those individuals who were homozygous for the GAA expansion the P and R wave axes were significantly different from those without an expansion, with minor inferior T wave changes being found in only one case compared with 50% of individuals who were homozygous for the GAA expansion who had widespread T wave inversion (table 3).
ECHOCARDIOGRAPHY
Cross sectional studies
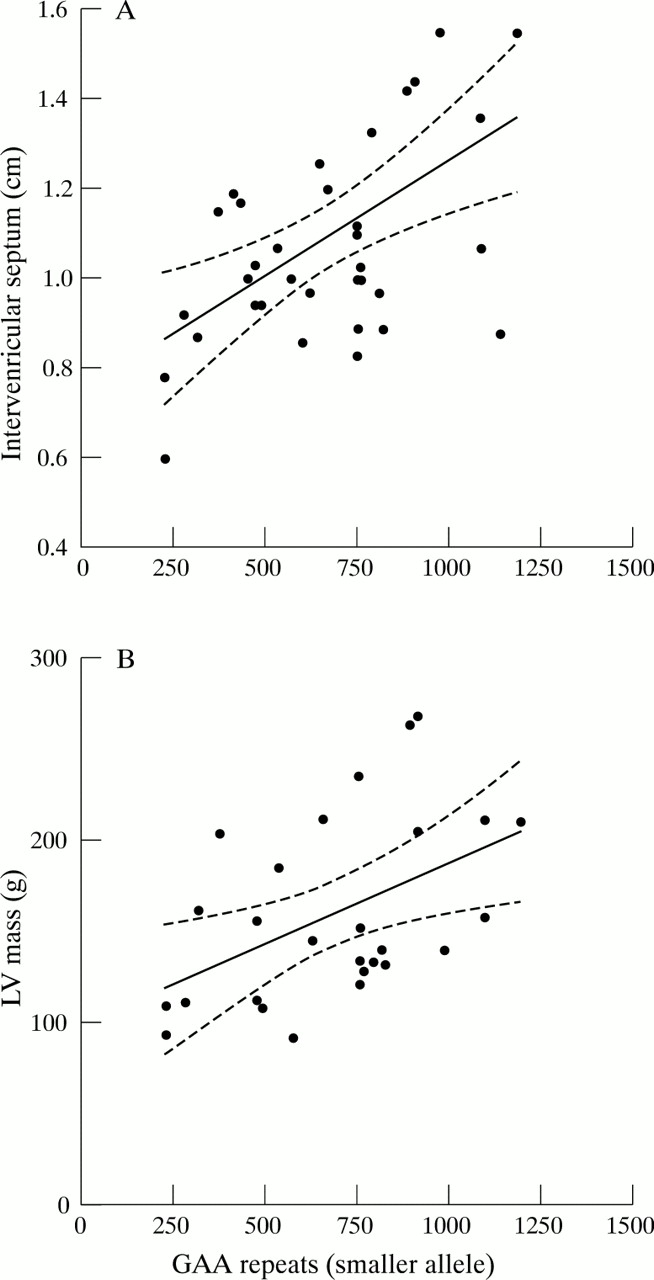
The echocardiogram was normal in 12 of the subjects studied with a clinical diagnosis of Friedreich’s ataxia, including all those with normal frataxin alleles (table 4). Left ventricular hypertrophy was present in 21 of those homozygous for the GAA expansion (fig 2). When present, the hypertrophy was confined to the proximal portion of the interventricular septum or posterior left ventricular wall. It was concentric in eight, but asymmetrical with a ventricular septum to posterior left ventricular wall ratio of > 1.2 in 13 of the subjects studied. A relation was found between an increasing number of GAA triplet repeats in the smaller frataxin allele and the thickness of the interventricular septum (r = 0.53, p < 0.005, fig 3A) and increasing left ventricular mass (r = 0.48, p < 0.01, fig 3B). Cardiac hypertrophy was not, however, related to the duration of disease. The left ventricle was dilated (internal diameter in diastole > 56 mm) in only one patient but impaired systolic function was documented in eight individuals. This was assessed independently by three of us as mild in five, moderate in two, and marked in the case where the ventricle was also dilated. In some subjects the left ventricle was hyperkinetic, with an echocardiographic left ventricular ejection fraction of > 70% in 15. Values were not corrected for surface area in view of the associated skeletal abnormalities and difficulty in establishing a correct value for the patient’s height.
Cross sectional echocardiographic features in Friedreich’s ataxia

Parasternal long axis echocardiogram of the patient whose ECG is shown in fig 1, illustrating hypertrophy of the proximal interventricular septum (14.2 mm) and normal dimensions of the left ventricle (LV) and atrium (LA), aorta (AO), and right ventricular outflow (RVO).
{kind=link}
{kind=link}
{kind=link}
Relation between (A) the thickness of the interventricular septum (in diastole, r = 0.53, p < 0.005), and (B) left ventricular (LV) mass (r = 0.48, p < 0.01) and the number of GAA repeats on the smaller allele of the frataxin gene in the 45 subjects homozygous for the GAA expansion. The 95% confidence intervals are shown by the dotted lines.
The papillary muscles were hypertrophied and prominent in eight subjects (who each had > 600 GAA repeats), associated with concentric hypertrophy in two and asymmetric hypertrophy in three. No evidence of systolic anterior motion of the mitral valve or valve prolapse was seen in any of the cases. The left atrium was not found to be significantly dilated in any of the subjects (maximum diameter 43 mm, mean left atrium to aorta ratio 1.33 (0.3)).
Doppler studies
Doppler studies were performed in all subjects (table 5). There was no relation between the Doppler indices of diastolic ventricular filling and the degree or pattern of hypertrophy. No subject was found to have a pressure gradient either within the left ventricle or in the outflow tract. However, aortic valve ejection time was shorter in subjects homozygous for the GAA expansion, with a lower peak velocity in the left ventricular outflow tract and a reduced velocity–time integral. These features all suggest left ventricular dysfunction although calculated ejection fraction was the same in the two groups (table 5).
Doppler findings in Friedreich’s ataxia
NEUROLOGICAL CORRELATION
No correlation was shown between the presence of ventricular hypertrophy and the age of onset of first symptoms (r 2 = 0.1), age at diagnosis of Friedreich’s ataxia (r 2 = 0.1), or age of confinement to a wheelchair (r 2 = 0.2). In addition there was no relation between the ability to walk and the presence of ventricular hypertrophy, and no difference in age between those with ventricular hypertrophy (29.5 (11) years) and those without (29.9 (11 years). Indeed, the three subjects with the most prominent electrocardiographic and echocardiographic abnormalities were still able to walk.
Discussion
We have shown in this prospective study that there is considerable variation in cardiac morphology in patients with a clinical diagnosis of Friedreich’s ataxia. The 55 individuals studied were not selected because of a history of cardiac symptoms but were respondents from a list of pedigrees where a clinical diagnosis of Friedreich’s ataxia had been made by a neurologist with an international reputation in the condition (the late professor A E Harding).
Symptoms referable to the heart were uncommon. Breathlessness was reported by 10 subjects, two of whom were confined to a wheelchair and both were noted to have impaired systolic function (one had undergone investigation including echocardiography and was receiving treatment with an ACE inhibitor). In the other eight subjects who were not yet confined to a wheelchair the breathlessness appeared to be related more to the considerable effort required to maintain balance and coordinate the lower limbs than to a cardiovascular cause. The only other reported symptom was an occasional awareness of the heartbeat which had prompted treatment with low dose β blockade in three cases without investigation.
The electrocardiogram was abnormal in the majority of subjects who were homozygous for the GAA expansion, abnormalities of ventricular repolarisation being the most common finding. In contrast, only one individual with normal frataxin alleles had minor T wave changes. The prevalence of these T wave changes was similar to that of previous reports,3 ,5 ,6 but an abnormal ECG is considered by some neurologists to be a prerequisite for the diagnosis of Friedreich’s ataxia. The diverse changes in the electrical axis of the heart remain unexplained although chest wall deformities may play a part. The abnormalities are clearly different from those seen in familial hypertrophic cardiomyopathy.
The echocardiographic studies revealed similar findings and the results were normal in all the subjects with normal frataxin alleles. The findings in those 45 individuals who were homozygous for the GAA expansion confirmed a diverse range of cardiac morphology. The absence of a relation between the pattern of hypertrophy (concentric or asymmetrical) and the age of onset of the first symptoms or age of dependence on a wheelchair confirms a discordance between the neurological changes and those seen in the heart. It has been suggested that the differing patterns of hypertrophy seen represent sampling along a continuum, documentation occurring at differing points in the natural history of the hypertrophic process.27 ,28 This hypothesis appears flawed for a number of reasons, particularly the absence of any relation between the neurological features (including age of onset and rate of progression of disability), the electrocardiographic findings, and the diverse pattern of cardiac morphology. Several reports have employed the term “hypertrophic cardiomyopathy”13 ,14 ,29 associated with Friedreich’s ataxia, but this description should be discarded. None of our subjects had the typical ECG appearances of the dominantly inherited familial hypertrophic cardiomyopathy, and the degree of hypertrophy of the interventricular septum in those individuals with asymmetrical septal hypertrophy was not gross (maximum 1.7 cm). In no case was a gradient noted in the left ventricle although some turbulence was seen associated with the marked hypertrophy of the papillary muscles when this was present. The possibility that some individuals with Friedreich’s ataxia might have familial hypertrophic cardiomyopathy as well is also remote; none of the families had a positive family history for premature sudden death, and the myocyte disarray and increased extracellular matrix seen in familial hypertrophic cardiomyopathy are not described in the heart in Friedreich’s ataxia.
Impaired left ventricular systolic function was present in a minority of cases, and marked in only one subject who had sought medical advice for persistent ankle oedema. In this case the ventricle was also dilated (diastolic diameter 6.0 cm), but the walls were not appreciably thinned (interventricular septum 1.0 cm and posterior wall of the left ventricle 0.9 cm) indicating that the total muscle mass was increased. In none of these subjects with reduced systolic function was there evidence of a regional wall motion abnormality suggestive of coronary artery disease. Intimal proliferation of small coronary vessels has been reported in Friedreich’s ataxia although this was not associated with ventricular dysfunction.3 One subject (with repolarisation changes on the ECG but a normal echocardiogram) was receiving insulin but none of the other individuals was known to have impaired glucose tolerance. It is very unlikely that occult diabetes or consequent coronary artery disease would have influenced the findings reported here as there was no evidence of regional wall motion abnormalities in any of the ventricles studied.
The report that Friedreich’s ataxia is secondary a GAA expansion in the first intron of the X25 gene, leading to reduced synthesis of frataxin, was based on findings in a group of patients who satisfied Harding’s diagnostic criteria.22 Ninety four per cent of these cases were homozygous for the triplet repeat expansion, with point mutations in heterozygotes appearing to confirm that frataxin exon 1 is an essential component of the protein which is defective in Friedreich’s ataxia. Dürr et al studied 187 patients with autosomal recessive ataxia and found that of the 140 who were homozygous for the GAA expansion in the frataxin gene, approximately 25% had atypical Friedreich’s ataxia.30Echocardiography was undertaken in 75 patients and cardiomyopathy was reported in 63% of patients who had a larger number of GAA repeats on the smaller allele, but there was no relation between the presence of cardiomyopathy and duration of disease. Similarly, in a study of 67 Italian patients with Friedreich’s ataxia, cardiomyopathy was reported in 25 of the 56 patients who underwent echocardiography, with significantly larger GAA expansions in those with cardiomyopathy.31 A relation between the number of GAA repeats on the smaller frataxin allele in patients with Friedreich’s ataxia homozygous for the GAA expansion and the thickness of the interventricular septum has also recently been reported in a group of 44 patients studied in Paris.32 However, all of these three studies were retrospective, the cardiac studies having been undertaken up to six years before the genetic analysis. These and other studies33 have confirmed that the clinical spectrum of Friedreich’s ataxia is broader than previously thought, and have led to the suggestion that Friedreich’s ataxia be redefined by excluding individuals without a GAA expansion in the frataxin gene. However, in the study by Dürr et al,3046% of patients with atypical Friedreich’s ataxia were homozygous for the expansion, although at least one essential clinical criterion was absent after five years of progressive, unremitting cerebellar ataxia.
Our finding of a relation between the size of the smaller GAA expansion and the thickness of the interventricular septum in individuals homozygous for this mutation in the frataxin gene is in accord with these previous reports, though the rather weak relation suggests that additional factors may be modifying phenotypic expression. The fact that some of the clinical variability is not related to the size of the GAA expansion has been highlighted by the findings in Acadian Friedreich’s ataxia. These patients have a distinct phenotype characterised by milder disease course and less severe cardiac involvement. A founder effect has been demonstrated, with most of the Friedreich’s ataxia chromosomes sharing a common core haplotype of markers flanking the disease locus. The phenotype of this variant would therefore appear to be linked to the Friedreich’s ataxia gene itself rather than to modifying genes present in the population. The GAA expansion is present in Acadian Friedreich’s ataxia, with the average expansion size similar to that of typical Friedreich’s ataxia. Polymorphic variation does not appear to account for these differences33 which supports the hypothesis that the clinical features associated with mutation of the frataxin gene are determined by several factors acting at the Friedreich’s ataxia locus.
Montermini and colleagues33 have proposed a revised clinical definition of Friedreich’s ataxia based on the molecular studies and excluding the rare isolated vitamin E deficient syndrome (AVED)34 which is not associated with cardiac abnormalities. Late onset and retained reflexes do not exclude the diagnosis, while the presence of cardiomyopathy invariably supports it (though the lack of cardiomyopathy does not exclude it).
Our study has highlighted the problems of redefining Friedreich’s ataxia on the basis of the triplet repeat expansion in the frataxin gene. In a study of 22 Irish families with typical Friedreich’s ataxia, 73% were homozygous for the expansion, 9% were heterozygous, and in 18% an expansion was not detected.35 If the presence of an expansion is used as the only diagnostic criterion there is a significant possibility of false negative results, as the clinical syndrome of Friedreich’s ataxia may be associated with the presence of a normal frataxin gene allele. Human frataxin has recently been identified as a mitochondrial protein regulating iron homeostasis, with iron deposition accounting for the biochemical and neurological lesions found in Friedreich’s ataxia.23 Studies on myocardial biopsy material from two subjects with Friedreich’s ataxia showed that frataxin gene mutations lead to deficiency of some Fe–S dependent respiratory enzyme chains, probably as a result of the oxidative stress caused by the iron overload in the mitochondrial matrix.24This requires confirmation in a larger group of patients, particularly the putative iron carrier system and its proposed regulation by frataxin. The differing antioxidant properties of human tissues such as the heart and particular neurones may contribute to the spectrum of clinical features seen in Friedreich’s ataxia, but this hypothesis does not account for the differing frequency of cardiomyopathy between typical Friedreich’s ataxia, the Acadian and retained reflex variants, and late onset Friedreich’s ataxia, where the size of the GAA mutation is similar. When data from all such subjects is considered together, the relation between the size of the GAA expansion and presence of cardiomyopathy is lost.33
It is difficult at present to construct a unifying aetiology for the cardiomyopathy associated with Friedreich’s ataxia. The role of frataxin in preventing mitochondrial oxidative damage in normal human myocardium requires further study, and the relation between mutated frataxin and other components of the mitochondrial respiratory chain needs to be explored to ensure that the spectrum of clinical features associated with Friedreich’s ataxia is not also related to variants in other genes.
CONCLUSIONS
Cardiac abnormalities are common in Friedreich’s ataxia and electrocardiographic abnormalities appear to be ubiquitous in individuals who are homozygous for trinucleotide expansion in intron 1 of the frataxin gene. However, the pattern of cardiac morphology is variable and is not simply related to the mutation in the frataxin gene or to the duration of neurological symptoms. We believe that there is no justification for amending the clinical definition of Friedreich’s ataxia until the molecular basis of the varying clinical features of the condition have been further defined. The condition offers a unique opportunity to further our understanding of myocardial hypertrophy in man.
Acknowledgments
This study was supported by the British Heart Foundation and the Special Trustees of the Hammersmith Hospital. We are grateful to Acuson (UK) Ltd for the loan evaluation of the echocardiographic equipment used in this study. We thank Dr Susan Chamberlain for her help.