Article Text

Abstract
Neisseria meningitidis is remarkable for the diversity of interactions that the bacterium has with the human host, ranging from asymptomatic nasopharyngeal colonisation affecting virtually all members of the population; through focal infections of the meninges, joints, or eye; to the devastating and often fatal syndrome of meningococcal septic shock and purpura fulminans.
- meningitis
- meningococcaemia
- septic shock
- CSF, cerebrospinal fluid
- IL, interleukin
- NCAM, neural cell adhesion molecule
- PAI, plasminogen activator inhibitor
- TFPI, tissue factor pathway inhibitor
- TNF, tumour necrosis factor
Statistics from Altmetric.com
- CSF, cerebrospinal fluid
- IL, interleukin
- NCAM, neural cell adhesion molecule
- PAI, plasminogen activator inhibitor
- TFPI, tissue factor pathway inhibitor
- TNF, tumour necrosis factor
In the past few decades, considerable progress has been made in understanding the complex interaction of host and pathogen, and the pathophysiology underlying both meningococcal septicaemia and meningitis. This increased understanding has resulted in improved management of the disorder, and is likely to lead to the introduction of new forms of treatment
HOST PATHOGEN INTERACTION: COLONISATION, INVASION, AND SURVIVAL IN THE BLOODSTREAM
The reasons why invasive disease occurs in a very small percentage of individuals are not well understood. A variety of factors including preceding viral infections, inhalation of dry dusty air, or exposure to passive smoking have been associated with invasive disease.1
The meningococcus is able to adhere to non-ciliated epithelial cells through a number of adhesion factors, including pili.2 The bacterium has a number of mechanisms enabling it to avoid host immunological mechanisms in the nasopharynx, including production of IgA protease,3 production of factors which inhibit ciliary activity,4 and the possession of a polysaccharide capsule which promotes adherence and inhibits opsonophagocytosis.5 The organism also undergoes notable variation in the level of expression or structure of a variety of surface antigens, including proteins and endotoxin, which enables the bacterium to evade the host immune response. In the case of group B meningococci, the polysaccharide structure is non-immunogenic, because of structural similarity with the human neural cell adhesion molecule (NCAM).6
While the events which enable certain strains of meningococci to pass through the nasopharyngeal membrane into the bloodstream remain poorly understood, it is clear that survival in the bloodstream, and the ability to multiply to high numbers, are an essential requirement for severe disease to occur.
The human immune system has a number of different mechanisms to limit the growth of meningococci in blood. Both the older innate immune system and acquired immunity through antibodies, T and B cells play a role. The epidemiology of meningococcal disease clearly indicates that the development of specific antibody is the most important immunoprotective mechanism.7,8 The peak incidence of the disease occurs in the first year of life following the loss of maternal antibody; the disease becomes progressively less common throughout childhood and is extremely rare in adults, a pattern suggestive of a major role for acquired immunity through antibodies. The studies of Goldschneider and Gottlieb established that the age related incidence of the disease in the population is related to the presence of bactericidal antibodies in serum.7 Although antibody appears to be the primary protective mechanism, there is also evidence that innate immune mechanisms are important in protecting infants, particularly in the window of time between colonisation of the nasopharynx and the development of protective immune response. All three components of the complement pathway—classical pathway through antibody; alternative pathway through properdin; and the newly discovered mannose binding lectin pathway—are important in the protection against meningococcal disease. Individuals deficient in terminal components of the complement pathway, as well as those with properdin deficiency, suffer recurrent infection.9,10 Individuals with mutations in the mannose binding lectin gene have an increased risk of meningococcal disease but appear less likely to suffer recurrent infections once antibodies have developed.11
ACTIVATION OF THE HOST IMMUNE RESPONSE
A large body of evidence now indicates that much of the damage to host tissues is caused by activation of host immune mechanisms (fig 1). Although there are likely to be numerous bacterial factors that can trigger the host immune response, endotoxin appears to be the most important. The studies of Brandtzaeg et al clearly established that the severity of meningococcal septicaemia was directly related to levels of circulating endotoxin.12 The characteristic property of meningococci to release blebs of outer membrane vesicles, which are rich in endotoxin, probably plays an important part in releasing large quantities of endotoxin into the bloodstream.13

The inflammatory cascade in meningococcal septicaemia.
Endotoxin is bound to a circulating plasma protein called endotoxin binding protein. The interaction between endotoxin and endotoxin binding protein alters the conformation of endotoxin to enable increased binding to and activation of macrophages and other inflammatory cells.14 The principle cellular receptor for endotoxin is CD14, but a range of other endotoxin sensors on cell surfaces exist, including the toll like receptors, which are also important in activation of host inflammatory cells.15,16 A soluble form of CD14 also acts as the receptor for endotoxin on endothelial surfaces. Endotoxin binding to these cells triggers an intense inflammatory process. Macrophages undergo activation to produce a range of proinflammatory cytokines, including tumour necrosis factor α (TNFα) and interleukin 1β (IL-1β). Increased production of interferon γ by T cells and natural killer cells occurs, and the interferon γ in turn enhances production of TNF by the macrophage. A number of studies have documented high levels of cytokines including TNFα, IL-1β, IL-6, IL-8, GM-CSF, IL-10, and interferon γ in meningococcal sepsis.17–19 In general cytokine levels are closely correlated with disease severity and risk of death.
Neutrophils appear to be activated both through the effects of endotoxin and through complement mediated stimuli. Neutrophils undergo a respiratory burst with the production of reactive oxygen species, as well as undergoing degranulation and the release of a range of inflammatory proteins, proteases, and other enzymes, which can degrade tissues. Close proximity and attachment of neutrophils to the endothelial lining in sepsis probably enable proteolytic enzymes to be released, which damage the endothelial surface.20
MICROVASCULAR INJURY IN MENINGOCOCCAL SEPSIS
There is now considerable evidence that indicates that the major pathophysiological event occurring in meningococcal septicaemia, which explains most of the severe physiological consequences of the disease, is related to a profound change in the normal finely regulated functions of the microvasculature. The vascular endothelial surface is a highly specialised organ, regulating vascular permeability and presenting a thromboresistant, non-reactive surface to circulating blood cells. These highly specialised properties are lost during the inflammatory process, occurring on entry of meningococci into the bloodstream. The complex physiology of meningococcal sepsis is largely explained by four basic processes affecting the microvasculature:
-
Increased vascular permeability
-
Pathological vasoconstriction and vasodilatation
-
Loss of thromboresistance and intravascular coagulation
-
Profound myocardial dysfunction.
These events are largely responsible for the development of shock and multiorgan failure.
Increased vascular permeability and the capillary leak syndrome
Hypovolaemia appears to be the most important early event leading to shock and is a direct result of a gross increase in vascular permeability. The inflammatory process induced by meningococci results in a major change in the permeability properties of the endothelium in all vascular beds. The increased permeability to plasma proteins results in proteinuria, similar in magnitude to the urinary albumin loss in children with steroid responsive nephrotic syndrome.21 Enzymatic degradation of endothelial surface proteins and loss of the surface endothelial glycosaminoglycans has been implicated in the aetiology of this capillary leak.21,22 Loss of albumin is followed by loss of fluid and electrolytes, and profound hypovolaemia ensues. Loss of circulating plasma is initially compensated for by homoeostatic mechanisms, including vasoconstriction of both arterial and venous vascular beds. However, as the capillary leak progresses, venous return to the heart is impaired and the cardiac output falls notably. Although restoration of circulation volume is the single most important component of resuscitation, it is also associated with the risk of increasing oedema within all tissues and organs as a result of persistent capillary leak. In addition to gross swelling of all the tissues and muscle compartments, high protein fluids may accumulate in the peritoneal and pleural spaces, and intra-alveolar space. Pulmonary oedema and respiratory failure are thus direct consequences of the gross increase in vascular permeability.
Pathological vasoconstriction and vasodilatation
Compensatory vasoconstriction is an important early protective mechanism to maintain tissue and organ perfusion in the face of diminished cardiac output. Most patients with meningococcal septic shock have evidence of intense vasoconstriction on admission, with cold peripheries and sluggish blood flows to the tissues. Although the vasoconstriction is primarily protective, the vasospasm may persist, even after resuscitation and measures to improve cardiac output. Severely affected patients may develop cold, pale, and ischaemic limbs. If this intense vasoconstriction persists, thrombosis within the microvasculature and gangrene inevitably results. Conversely some patients have intense vasodilatation following resuscitation, with bounding pulses, warm periphery, and yet severe hypotension, acidosis, and organ impairment, the classical picture of so-called “warm shock”. Between these two extremes of vasomotor response there are other patients who have a mixture of profound vasoconstriction of some vascular beds and dilatation of others.
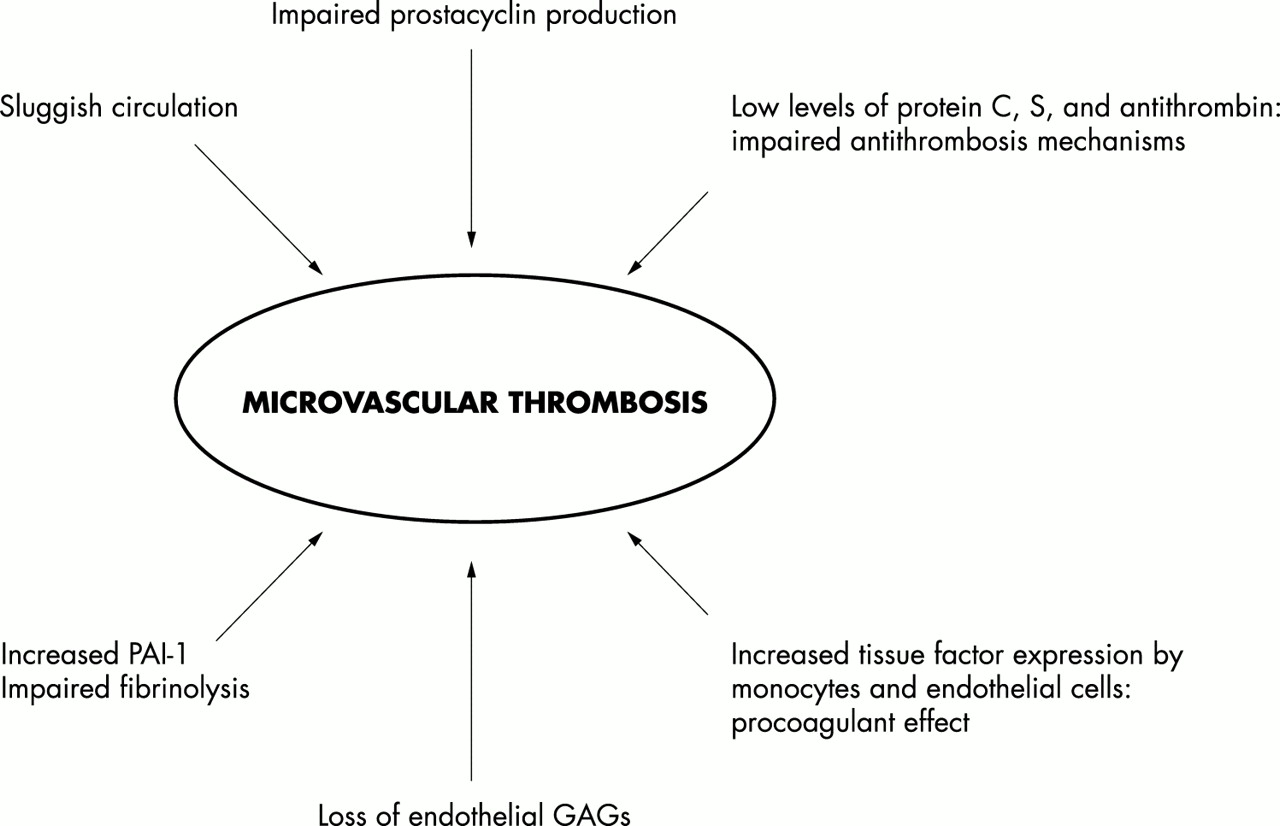
Intravascular thrombosis
One of the most dramatic features of severe meningococcal sepsis is the occurrence of widespread purpura fulminans,23 with thrombosis and haemorrhagic necrosis in large areas of skin and in extreme cases with infarction and gangrene of limbs and digits. The severe disseminated intravascular thrombosis occurs in the context of profound thrombocytopenia and prolonged coagulation.24 Normal vascular homoeostasis involves carefully regulated interaction of procoagulant, antithrombotic, and thrombolytic pathways (figs 2 and 3). Procoagulant pathways are activated in meningococcal sepsis, and there is impairment of both the natural anticoagulant pathways and the fibrinolytic system. It remains unclear why purpura fulminans develops in some patients, whereas in others with equally severe septic shock there are no thrombotic complications. The endothelium plays an important role in thromboresistance. The presence of heparin like molecules such as heparan sulphate on the endothelium is an important antithrombotic mechanism.22,25 The endothelium also produces prostacyclin and nitric oxide, which inhibit platelet activation.

(A) The inflammatory, coagulation, and fibrinolytic pathways are linked at many levels, leading to organ failure and eventually death. (B) The coagulation and fibrinolytic pathways rely on the function of endothelial proteins and protein complexes. Dysfunctional regulatory mechanisms in meningococcal disease include endothelial (protein C pathway) and plasma (TFPI, antithrombin, PAI) factors.

{kind=link}
{kind=link}
{kind=link}
Factors involved in intravascular thrombosis and purpura fulminans.
The endothelium plays a major role in the activation of the protein C pathway. Protein C is a vitamin K dependent plasma protein that circulates in plasma as an inactive zymogen. Once activated, protein C requires protein S as a cofactor for its anticoagulant functions.26 In sepsis, upregulation of procoagulant pathways leads to intravascular generation of thrombin.27,28 Thrombin normally binds to thrombomodulin on the endothelial surface and subsequently binds protein C. This enables protein C activation, which acts with protein S, inactivating factor Va and factor VIIIa, as well as downregulating plasminogen activator inhibitor. Virtually all these antithrombotic mechanisms appear to be dysfunctional in meningococcal sepsis.29,30 Prostacyclin production by endothelium is impaired on incubation with plasma from children with meningococcal sepsis.31 The binding and activity of antithrombin on endothelial surfaces is downregulated after exposure to meningococci and endotoxin in vitro.25 In vivo studies on skin biopsy samples have documented loss of both thrombomodulin and endothelial protein C receptor in meningococcal sepsis.32 The plasma levels for the anticoagulant proteins antithrombin, protein C, and protein S are all reduced in meningococcal sepsis and the generation of activated protein C is profoundly impaired, probably as a consequence of loss of endothelial thrombomodulin and endothelial protein C receptors.32 Furthermore, levels of tissue factor pathway inhibitor (TFPI) are reduced in sepsis. Loss of these anticoagulant mechanisms, together with upregulation of tissue factor on monocytes (and presumably on endothelium), and the release of procoagulant molecules from platelets and monocytes, triggers uncontrolled intravascular coagulation.33 Thrombolytic mechanisms also appear to be profoundly impaired. Both plasminogen activator, an initiator of the thrombolytic pathway, and its inhibitor, plasminogen activator inhibitor 1 (PAI-1), are released during meningococcal induced inflammatory stimuli.34 Levels of PAI-1 are profoundly increased in meningococcal sepsis and show a direct correlation with the severity of the disease.35 Moreover, a genetic polymorphism in the PAI-1 promoter, which favours high levels of production of plasminogen activation inhibitor is associated with increased risk of death.36 High levels of PAI-1 impair thrombolytic activity, and together with profound defects in antithrombotic mechanisms, leads the way to widespread intravascular thrombosis. Furthermore, sluggish capillary circulation, intense vasoconstriction which attempts to compensate for diminished cardiac output, increases the likelihood of venous thrombosis.
Myocardial dysfunction in meningococcal sepsis
Septicaemia is associated with notable changes in haemodynamic load and acute myocardial failure.37 In mild cases this may manifest only as compensated shock with tachycardia and vasoconstriction, but in severe cases hypotension and severe impairment of tissues and organs occurs. Hypovolaemia caused by the capillary leak and loss of circulating volume is undoubtedly a major contributor to the myocardial dysfunction in sepsis. However, there is also evidence of a profound defect in myocardial contractility, which persists even after correction of circulating volume. Haemodynamic studies have shown that the severity of disease in meningococcal sepsis is related to the degree of impairment of cardiac contractility.38 Precise mechanisms responsible for impaired myocardial contractility are as yet unclear. The development of myocardial dysfunction is thought to be caused by the negatively inotropic effect of various proinflammatory mediators released in sepsis. A variety of proinflammatory substances released during septic shock are known to depress myocardial function. These include nitric oxide, TNFα and IL-1β,39,40 and possibly other as yet not fully defined myocardial depressant factors: serum from children with meningococcal disease depresses myocardial function in vitro, and is not due to either TNFα or IL-1β, but appears to be caused by other proinflammatory mediators.41 Furthermore, hypoxia, acidosis, hypoglycaemia, hypokolaemia, hypocalcaemia, and hypophosphataemia are all common in severe meningococcal sepsis and may adversely affect myocardial function. Some patients appear to become unresponsive to the positive inotropic effects of catecholamines, and exhibit increasing requirements for therapeutic inotropes. Although the impaired myocardial contractility generally improves once hypovolaemia, acidosis, and electrolyte imbalance have been corrected, in some patients prolonged impairment of myocardial contractility occurs. The myocardial impairment appears to be reversible in most patients, although there is now evidence that cardiac troponin I—a recognised marker of myocardial damage—is increased in meningococcal septicaemia in relation to severity of the disease.42 Some long term damage to the myocardium may thus occur in severely affected patients.
IMPAIRED ORGAN PERFUSION
The four processes described above result in impairment of microvascular blood flow to the tissues and organs of the body.
RENAL IMPAIRMENT
Most patients with meningococcal sepsis show evidence of impaired renal perfusion, which is related to the severity of shock.12 If the impaired renal perfusion persists, patients become oligoanuric with a rise in serum urea and creatinine.37 In most patients the impairment of renal function is transient; urine output increases once volume resuscitation and improved cardiac output has been achieved. However, in severe cases progression to vasomotor nephropathy and acute tubular necrosis develops.
PULMONARY INVOLVEMENT
Pulmonary function is affected very early by the capillary leak occurring in meningococcal sepsis. Tachypnoea and increased work of breathing occur, and in some patients hypoxic respiratory failure is present almost immediately on admission. Volume resuscitation and expansion of circulating volume inevitably results in increased intra-alveolar fluid if the capillary leak is severe, leading to pulmonary oedema and respiratory failure. Although little is known about the microvascular events occurring in the lung in meningococcal sepsis, it is likely that the widespread adhesion of neutrophils to the endothelium, activation of coagulation, and activation of platelets which is occurring throughout this vascular bed is also occurring within the lung, and may result in microvascular obstruction.43
GASTROINTESTINAL CONSEQUENCES
Reduced blood flow to the gastrointestinal tract occurs early in the course of septic shock. Most patients’ gastrointestinal function improves once cardiac output is increased. However, in severe cases evidence of profound gastric ischaemia may be seen with prolonged ileus, and occasionally with ischaemic ulceration and perforation occurring later on in the disease.44
CENTRAL NERVOUS SYSTEM INVOLVEMENT IN SEPTIC SHOCK AND MENINGITIS
Impaired central nervous system function may be seen in meningococcal disease as a result of both direct invasion of the meninges by the bacteria and as part of the organ under perfusion in septic shock. Many patients have a mixed picture with evidence of an inflammatory process triggered by the bacteria within the cerebrospinal fluid (CSF), as well as impaired neurological function to profound shock.
There are important clinical distinctions between raised intracranial pressure and brain inflammation occurring as part of meningitis, and brain and central nervous system malfunction occurring as part of meningococcal septicaemia and septic shock. A significant proportion of children with meningococcal meningitis develop raised intracranial pressure as a consequence of the inflammatory process within the brain. In these patients rapid development of raised intracranial pressure may occur, which in extreme cases may result in cerebral herniation. Unfortunately the clinical features of patients with raised intracranial pressure caused by meningitis may overlap with those with severe central nervous system dysfunction during the course of meningococcal sepsis. Profoundly shocked patients invariably show diminished consciousness and are at risk of cerebral infarction if perfusion is not improved.
It is likely that distinct pathophysiological events underlie cerebral oedema and raised intracranial pressure seen in those patients with purely meningitis, as opposed to those with neurological dysfunction caused by underperfusion and septic shock. Neurological damage in meningitis may be caused by a mixture of direct bacterial toxicity, indirect inflammatory processes such as cytokine release, neutrophil activation, with resultant vasculitis, and cellular oedema.45,46 Cerebral oedema may be caused by increased secretion of CSF, diminished reabsorption of CSF, and breakdown of the blood-brain barrier.47 In contrast the neurological dysfunction in shock results from reduced perfusion and microvascular obstruction, which may lead to cerebral infarction.
THERAPEUTIC IMPLICATIONS
Our understanding of the pathophysiology of meningococcal septic shock allows a rational approach to treatment to be developed and are described in detail by Welch and Nadel.48 Perhaps the most difficult area to define rational treatment is in measures to reduce the inflammatory process and prevent the widespread intravascular coagulation. A better understanding of the pathophysiology has led to a number of experimental therapies, targeting various inflammatory mediators (table 1). These include antiendotoxin agents such as bactericidal permeability increasing protein (rBPI21), which bind to endotoxin, preventing inflammatory cell activation; monoclonal antibodies against endotoxin; agents which dampen the proinflammatory burst of immune cells, such as steroids; and agents that block or neutralise cytokines such as TNF or IL-1. A multicentre placebo controlled trial of rBPI21 in meningococcal sepsis suggested a benefit and improved outcome, although the trial was underpowered to detect an effect on mortality.49 Further evaluation of rBPI21 administered earlier in the course of the disease is now being considered. Activated protein C has been shown to reduce mortality in adult sepsis,50 and placebo controlled trials of this agent in childhood sepsis are commencing in the near future. The growing understanding of the inflammatory process involving the introduction of anti-inflammatory treatments into clinical use in the future now seems likely.
Immunomodulation of sepsis: summary of experimental and clinical studies