Article Text

Abstract
BACKGROUND Learning disability and short stature are cardinal signs of Down’s syndrome. Insulin-like growth factor I (IGF-I), regulated by growth hormone (GH) from about 6 months of age, may be involved in brain development.
AIMS To study long term effects of GH on linear growth and psychomotor development in young children with Down’s syndrome.
Study design—Fifteen children with Down’s syndrome were treated with GH for three years from the age of 6 to 9 months (mean, 7.4). Linear growth, psychomotor development, skeletal maturation, serum concentrations of IGF-I and its binding proteins (BPs), and cerebrospinal fluid (CSF) concentrations of IGF-II were studied.
RESULTS The mean height of the study group increased from −1.8 to −0.8 SDS (Swedish standard) during treatment, whereas that of a Down’s syndrome control group fell from −1.7 to −2.2 SDS. Growth velocity declined after treatment stopped. Head growth did not accelerate during treatment. No significant difference in mental or gross motor development was found. The low concentrations of serum IGF-I and IGFBP-3 became normal during GH treatment.
CONCLUSIONS GH treatment results in normal growth velocity in Down’s syndrome but does not affect head circumference or mental or gross motor development. Growth velocity declines after treatment stops.
- Down’s syndrome
- growth retardation
- growth hormone treatment
- mental development
Statistics from Altmetric.com
Down’s syndrome, the most common single specific cause of learning disability, is characterised by short stature. Growth velocity is most reduced between the ages of 6 months and 3 years but subsequently is almost normal.1 ,2
In normal children, endogenous growth hormone (GH) has a major influence on growth from the age of 6 to 9 months, through stimulation of the production of insulin-like growth factor I (IGF-I).2 Thus, the growth retardation in Down’s syndrome becomes pronounced during the period when GH starts to regulate growth. There is no obvious deficiency of GH in the serum of children with Down’s syndrome,3-5 although suboptimal endogenous GH production as a result of hypothalamic dysfunction has been demonstrated.6 Selective deficiency of IGF-I in the serum has been seen in patients with Down’s syndrome older than 2 years.2 Patients with Down’s syndrome have no deficiency of serum IGF-II,7and IGF receptors are present in brain cells from fetuses with trisomy 21.8 In a previous study, we observed that children aged 3–6 years with Down’s syndrome who were short responded to short term GH treatment with an increased growth velocity and normalisation of serum concentrations of IGF-I.5
In this trial, we studied the long term effects of GH treatment on linear growth and on the mental and motor development of young children with Down’s syndrome.
Materials and methods
PATIENTS
Initially, the study group consisted of 16 children (12 boys and four girls) with Down’s syndrome. One girl was excluded after 12 months of treatment because of increased aminotransferases. All the children were being cared for by their biological parents. One boy had congenital urethral stenosis, surgically corrected at the age of 13 months. He had normal renal function. Fifteen age matched children (six boys and nine girls) with Down’s syndrome served as a control group. No child in either group suffered from any congenital malformations of the heart. The trial was approved by the ethics committee of the University of Uppsala and by the Medical Products Agency of Sweden.
TREATMENT
The children were between 6 and 9 months of age (mean, 7.4) when GH treatment began. The children in the study group received daily injections of recombinant human GH (Genotropin, Pharmacia and Upjohn, Stockholm, Sweden) at a dose of 0.1 IU/kg body weight/day for three years from the age of 6 to 9 months (mean, 7.4).
MONITORING
The means of three measurements of all anthropometric parameters (supine height before 2 years of age and, thereafter, standing height, measured with a stadiometer, weight and head circumference) were obtained at each occasion. These measurements and video recordings of neurological examinations were made at the start of the trial, every third month during the first year, and every sixth month during the second and third years. The procedures were also performed 12 months after GH treatment stopped, and at 5 and 6 years of age. The mean heights and mean head circumferences of the study and control groups before and during treatment were plotted on standard growth charts for healthy Swedish children9 and growth charts for children with Down’s syndrome.1
Bone age was determined according to the methods of Greulich and Pyle (GP)10 and Tanner and Whitehouse (TW2),11 and tests of motor (motor perceptual test)12 and mental development (Griffith’s test)13 were performed before GH treatment, after one year of treatment, at the end of treatment, and one year after treatment was stopped.
BLOOD AND CSF ANALYSES
The following blood analyses were performed as part of the project or for the sake of safety before treatment started, every six months during treatment, and one year after it was stopped: thyroid stimulating hormone (TSH), free thyroxin (fT4), haemoglobin concentration, erythrocyte volume fraction (EVF), platelet count, differential leucocyte count, bilirubin, liver enzymes (aspartate aminotransferase, alanine aminotransferase, and alkaline phosphatase), creatinine, gliadin antibodies, IGF-I, and insulin-like growth factor binding proteins 1 and 3 (IGFBP-1 and IGFBP-3). In addition, five days after the start of GH treatment, serum samples for the assay of IGF-I, IGFBP-1, and IGFBP-3 were obtained. Before the start of treatment, after one year, at the end of treatment, and one year after its termination cerebrospinal fluid (CSF) samples were obtained for assay of IGF-II. In three patients CSF for this purpose was also obtained five days after the start of treatment.
RADIOIMMUNOASSAYS
Insulin growth factor I
Serum samples were acid/ethanol extracted before IGF-I radioimmunoassay (RIA) to partially separate IGFs from IGFBPs, and125I-labelled des(1–3)IGF-I radioligand was used in the RIA to avoid interference by IGFBPs not removed by the extraction procedure, as described previously.14 The recovery of cold IGF-I was 98% and the intra-assay and interassay coefficients of variation were 4% and 11%, respectively. The lowest detectable quantity of IGF-I was 0.02 ng/tube. Cross reaction was < 0.1% with insulin and < 2% with IGF-II. Age dependent IGF-I z scores were calculated as described by Juul et al.15
Insulin growth factor II
CSF concentrations of IGF-II were determined by IGF-II RIA16 after acid size exclusion chromatography to exclude interference of IGFBPs and prohormone forms of IGF-II. The lowest detectable quantity of IGF-II was 0.02 ng/tube. Crossreactivity with IGF-I and insulin was 0.1% and < 0.01%, respectively.
Acid size exclusion chromatography
CSF samples (500 μl) were lyophilised, dissolved in 1 M acetic acid, incubated for six hours at 4°C, and separated on a Sephadex G-50 column (1 × 100 cm) equilibrated with 0.1 M acetic acid. Fractions of 1 ml were collected in bovine serum albumin (BSA) coated tubes, lyophilised, and resuspended in 0.05 M sodium phosphate, 0.15 M NaCl, 0.02% sodium azide, and 0.5% RIA grade BSA, pH 7.4.
IGFBP-1
Serum IGFBP-1 was determined by RIA as described by Póvoa and co-workers.17 The intra-assay and interassay coefficients of variation were 3% and 11%, respectively, and the detection limit was 3.0 ng/ml. Cross reactivities with IGFBP-2 and IGFBP-3 were less than 0.5 and 0.05%, respectively.
IGFBP-3
Serum IGFBP-3 was determined by a modified version of an RIA described earlier.18 Briefly, rabbit serum against glycosylated IGFBP-3 (a-IGFBP-3-g1; 1/1000 dilution; 50 μl), serum samples (1/200 dilution; 50 μl), or a reference sample of glycosylated IGFBP-3 and 125I-labelled IGFBP-3 (6000 counts/min) were incubated for 18 hours at 4°C in a total volume of 250 μl of assay buffer (0.05 M sodium phosphate, 0.15 M NaCl, 0.02% sodium azide, and 0.05% RIA grade BSA, pH 7.4). Antibody bound and free tracers were separated by centrifugation after incubation with Sepharose coupled second antibodies (Pharmacia Diagnostics AB, Uppsala, Sweden). Pellets were counted in a gamma counter. The intra-assay and interassay coefficients of variation were both < 9.3% (B/B0 = 80%), 4.5% (B/B0 = 30%) and 7.8% (B/B0 = 15%), respectively. The detection limit was 0.03 ng/tube. The cross reactivity with purified human IGFBP-1, IGFBP-2, or IGFBP-4 was < 0.3%.
STATISTICAL ANALYSIS
The two tailed Student’s t test was used. The values are presented as mean (SEM) or (SD).
Results
During the trial, one girl developed coeliac disease and she was treated with a gluten free diet. In another girl, GH treatment was stopped after one year because of raised serum concentrations of aminotransferases. She was excluded from analysis. No other child displayed any abnormalities in blood chemistry or morphology during treatment. All children tolerated the injections well and no side effects were noted. Fewer than one injection each month was forgotten or not given for other reasons, according to parents.
Before GH treatment, the study group had a mean (range) height standard deviation score (SDS; Swedish standard) of −1.8 (−0.5 to −3.1) (fig 1A). After 36 months of treatment this score had increased to −0.8 (−0.5 to −2.0). The corresponding figures for the control children were −1.7 (0.5 to −4.0) (not significant) and −2.2 (−0.8 to −4.0; p < 0.001), respectively (fig 1A). During GH treatment the growth rate in the children with Down’s syndrome followed that of healthy Swedish children. In fig 1B the mean growth of the study and control groups are presented in syndrome specific growth charts for children with Down’s syndrome.1 The mean height of both groups started at the 50th centile. Both the study and control groups displayed better growth than average children with Down’s syndrome. The mean bone age (GP and TW2) was somewhat retarded at the start of GH treatment, but was in the normal range after three years of treatment (fig 2).

Mean height of 12 boys and three girls with Down’s syndrome treated with growth hormone (GH) for three years from the age of 6–9 months (mean, 7.4), compared with that of an untreated group of six boys and nine girls with Down’s syndrome. (A) The results are presented on the Swedish growth chart for normal boys.9 (B) The results are presented on the growth chart for boys with Down’s syndrome.1

Mean (SEM) bone age, calculated according to Greulich and Pyle (GP), and Tanner and Whitehouse (TW2), in relation to chronological age in 15 children with Down’s syndrome treated with growth hormone for three years.
The mean (range) IGF-I SDS15 of the study group was −1.6 (−0.24 to −2.47) before GH treatment, 0.28 (−0.90 to 2.16) after 3 years of treatment, and −1.11 (0.15 to −1.86) one year after the end of treatment. The mean serum concentrations of IGF-I (fig 3A) and IGFBP-3 (fig 3B) increased rapidly after the start of GH treatment and continued to rise during the treatment period. During the year after the end of treatment both decreased (fig 3A and B). The mean serum concentration of IGFBP-1 (fasting morning sample) was in the normal range and did not change during GH treatment. Nor did the treatment have any effect on the mean concentration of IGF-II in CSF (fig 3C).
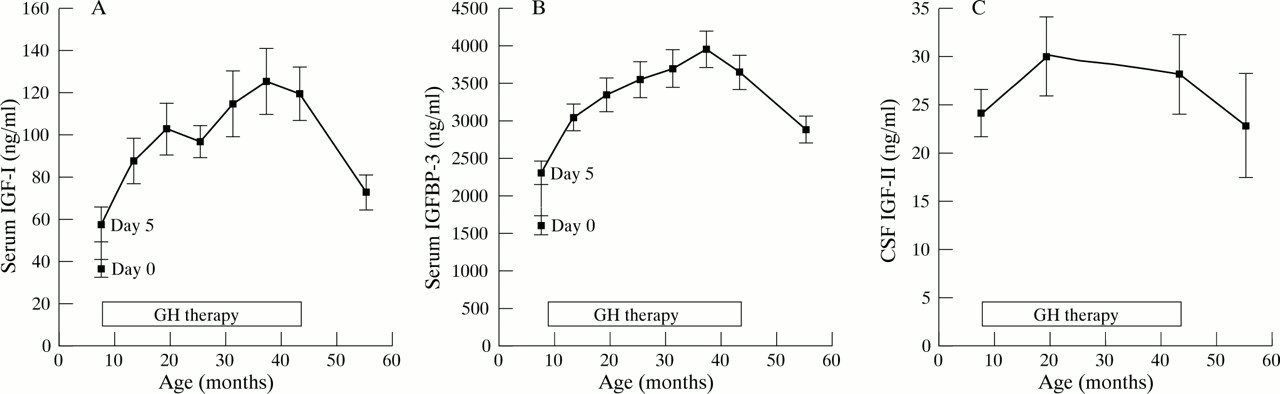
(A) Mean (SEM) serum concentrations of insulin-like growth factor 1 (IGF-I) in 15 children with Down’s syndrome treated with growth hormone (GH) for three years from the age of 6 to 9 months (mean, 7.4). (B) Mean (SEM) serum concentrations of IGF binding protein 3 (IGFBP-3) in 15 children with Down’s syndrome treated with growth hormone for three years from the age of 6 to 9 months (mean, 7.4). (C) Mean (SEM) concentrations of IGF-II in the cerebrospinal fluid (CSF) in 15 children with Down’s syndrome treated with growth hormone for three years from the age of 6 to 9 months (mean, 7.4).
There was a difference in head circumference between the two groups at the start of our study, and this difference did not change during the period of treatment. The mean (range) head circumference SDS in the study group was −1.4 (1.2 to −4.0) before and −1.2 (0.0 to −3.0) after treatment. The corresponding figures in the controls were −1.9 (0.0 to −5.0) and −2.0 (0.0 to −3.0), respectively.
After the end of GH treatment the mean height of the study group was significantly above that of the control children, but during the following years the study group grew less than during the treatment period and more slowly than the control children. However, at the age of 6.5 years (three years after the end of treatment) there was still some difference in height between the groups (p < 0.05) (figs 1A and B).
The mean IQ (Griffith’s test) decreased with age in the GH treated group (fig 4). No differences concerning mental development (Griffith’s mental development scales) or gross motor development were observed between the control and treated groups at the age of 3.5 years—when the GH treatment was ended. However, a somewhat better fine motor performance (developmental quotient 73 v60.5; p < 0.01) was noted in the GH treated children.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mean (SEM) mental development (IQ scores; Griffith’s mental development scale) from the age of 6 to 9 months to 4.5 years in 15 children with Down’s syndrome treated with growth hormone for three years.
Discussion
Growth velocity in children with Down’s syndrome is greatly reduced between the ages of 6 and 9 months and 3 years.1 ,2 During these periods, learning disability also becomes obvious and there is a decline in the mean intelligence quotient from 70 at the age of 12 months to 40–50 at 3 years.19 Subsequently, there is only a slight further decline in IQ.20 For this reason, we chose to start GH treatment at the age of 6–9 months to see whether it could prevent retardation of growth and mental development.
Our results have shown that GH eliminates the marked reduction of growth velocity in children with Down’s syndrome occurring between the ages of 6 months and 3 years. This is clearly evident when growth is plotted on the standard growth chart of children with Down’s syndrome.1 Thus, after 24 months of treatment all GH treated children were above the 95th centile for children with Down’s syndrome. It should be mentioned that a slightly increased growth velocity was also seen in the control children compared with the Down’s syndrome standard.1 This could be explained either by the fact that none of the children had any congenital heart disease or that the study group represented a Swedish population. During treatment we observed no acceleration of bone age, but because the bone ages of children with Down’s syndrome at the ages of 3.5 and 4.5 years have been reported to be retarded by about one year19 we cannot rule out a possible effect on skeletal maturation in the study group. Although there was a distinct effect on linear growth velocity during GH treatment, we cannot predict the influence of treatment on final height. A marked “catch down” effect on linear growth was seen during the year after treatment stopped. Thus, growth hormone treatment probably has to be continued for longer periods or until the end of linear growth to achieve a beneficial effect on final height.
The children tolerated the GH injections well. In one girl, the treatment was stopped after one year because of slightly raised serum concentrations of aminotransferases. We have observed transient increases in serum aminotransferases in children with Down’s syndrome not receiving GH treatment, and it is questionable whether the changes noted were related to the treatment. Coeliac disease with no negative effect on growth or weight gain was diagnosed in one girl at the age of 1 year, and she was treated with a gluten free diet. Because individuals with Down’s syndrome are at increased risk of developing acute leukaemia,21 repeated investigations of blood cell morphology were performed, but no abnormalities in this respect were found during the treatment period or in the following years. Parents failed to give fewer than one injection each month.
The fasting concentration of IGFBP-1 has been shown to correlate with the mean 24 hour concentration of IGFBP-1 and to reflect preceding diurnal insulin secretion.22 Thus, the observation of unchanged, normal fasting IGFBP-1 concentrations during the study period points to normal insulin action during the period of treatment.
GH treatment had no effect on head circumference, a finding in contrast to that of Torrado et al,23 who reported a significant increase in head growth velocity during treatment with GH. The growth of the central nervous system (CNS) has not been reported to be related to GH, although it is well documented that IGFs are important brain growth promoting hormones.24 IGF-I and IGF-II are known to stimulate nerve and glial cell proliferation and differentiation via the IGF-I receptor.25 Both IGF-I and IGF-II have local action in the brain, but only IGF-II is detectable in CSF.24 IGF-I may have a neuroprotective role and it has been suggested as a therapeutic agent for neurodegeneration.26 The question of whether endogenous GH has effects upon mental development has not been settled. Although children with GH deficiency are of normal intelligence, adults with GH deficiency have been reported to improve their psychological capacity after GH treatment.27 Thus, GH treatment in Down’s syndrome is of potential interest, although no information is available as to whether the local production of IGF-I in the brain is disturbed in Down’s syndrome and whether such a disturbance could contribute to brain dysfunction.24 Furthermore, despite the presence of brain growth hormone receptors,28 it is not known whether peripherally administered GH is able to reach the CNS nerve or glial cells. Because no stimulatory effect of exogenous GH on peripheral IGF-II production has been reported,29 the absence of changes in the IGF-II concentration in the CSF following GH treatment in our study does not provide any information as to whether exogenous GH reaches the brain or not.
In our study, we observed no effect on mental or motor development, except for some improvement of fine motor skill. Theoretically, this effect would be compatible with the report of a high density of IGF-I receptors in the cerebellum.30 To establish whether GH treatment really influences fine motor skill a larger study and control group would be required.
At present, we and others do not recommend GH treatment of children with Down’s syndrome without proven GH deficiency.31 ,32Although this treatment has a definite effect on linear growth, long term studies on larger groups of children are needed before routine treatment can be considered with the aim of improving final height in short stature children with Down’s syndrome. There is no documented relation between GH treatment and leukaemia,33 but future studies should focus on the possible occurrence of such adverse events in this particular group of children.
Acknowledgments
The skilful assistance of M Elfvik-Strömberg, E Enger, and B Westerberg is gratefully acknowledged. We are grateful to R Rosenfeld for supplying a-IGFBP-3-gl. This study was supported by the First of May Flower Foundation, the Sven Jerring Foundation, Pharmacia and Upjohn (Stockholm, Sweden), the Sävstaholm Foundation, the Gillberg Foundation, the Sven Johansson Foundation, and the Swedish Medical Research Council (grants 5445, 6446, 11634, and 2604).