Article Text

Abstract
Giant cell hepatitis (CGH) with autoimmune haemolytic anaemia (AHA) is a distinct entity with an aggressive course. Immunosuppression may help early disease. A case is reported of a child with GCH and AHA with early disease recurrence after liver transplantation for end stage liver disease.
- liver transplantation
- giant cell hepatitis
Statistics from Altmetric.com
Giant cell hepatitis (CGH) with autoimmune haemolytic anaemia (AHA) is a rare entity, distinct from other diseases with giant cell transformation, which include neonatal and postinfantile or syncytial GCH. GCH with AHA has an aggressive natural history leading to hepatic failure and death,1-3 although a response to immunosuppressive treatment has been reported.1 3Orthotopic liver transplantation (OLT) has been used successfully to treat other forms of giant cell transformation,4 5 and has been proposed for this condition.6
Case report
A 14 month old boy was referred for investigation. He was an only child born by forceps delivery after a normal 42 week pregnancy and had mild neonatal jaundice. At 8 months of age, he was admitted with failure to thrive, vomiting, and a chest infection and was found to have hepatosplenomegaly. Full blood count was normal, but a Coombs test was positive and a diagnosis of AHA was made. The serum aspartate aminotransferase rose to 6000 IU/l with a total bilirubin of 260 μmol/l.
On referral, the serum aspartate aminotransferase was 795 IU/l, bilirubin 232 μmol/l, and haemoglobin 110 g/l. A chest radiograph showed extensive bilateral hilar shadowing. Tests for tissue autoantibodies, hepatitis A, B, and C, Epstein-Barr virus, cytomegalovirus, HIV, herpes simplex virus, adenovirus, toxoplasmosis, and mycoplasma were negative. A liver biopsy specimen showed GCH with necrosis and collapse. GCH with AHA was diagnosed, and he was treated with prednisolone (2 mg/kg/day) and azathioprine (0.25 mg/kg/day), after an eight day course of methylprednisolone (16 mg/day). The aspartate aminotransferase rose to 7450 IU/l, but gradually improved over three weeks and he was discharged on azathioprine (8 mg/day) and prednisolone (10 mg/day).
Three months later, a rise in alanine aminotransferase to 551 IU/l led to the introduction of cyclosporin A (6 mg/kg/day) in place of azathioprine, but the serum aspartate aminotransferase rose to 1260 IU/l with haemolysis needing blood transfusion. Cyclosporin was replaced by penicillamine at gradually increasing doses up to 20 mg/kg/day, over six weeks with some improvement. Three months later he was readmitted with anaemia, ascites, jaundice, fever, and a grand mal convulsion. No cause was identified for the grand mal convulsion, and he was treated with aciclovir, phenytoin, and carbamazepine. Because of worsening liver function he was listed for OLT.
At 28 months of age, he was transplanted with a sized matched graft and received tacrolimus immunosuppression. The liver had a micronodular cirrhosis with evidence of marked portal hypertension. Histologically, there were areas of multiacinar collapse with occasional islands of less damaged tissue, numerous multinucleated giant hepatocytes, widespread cellular cholestasis, and mild mixed inflammatory cell infiltration (fig 1A and 1B). A liver biopsy specimen one week after transplant showed minimal rejection and the next week he was discharged on prednisolone (7.5 mg/day), azathioprine (16.5 mg/day), and tacrolimus (16 mg/day). Five weeks after transplant, a liver biopsy specimen showed evidence of perivenular injury with mild cell drop out and occasional multinucleated hepatocytes suggesting recurrent disease. He developed further grand mal fits, treated with clonazepam (2 mg/day). An electroencephalogram showed epileptogenic foci in both hemispheres. Intravenous immunoglobulin (400 mg/kg/day) was given for four days each month, without improvement. A further biopsy specimen showed severe perivenular damage associated with prominent, pigment laden, multinucleated giant cells and no rejection. The aspartate aminotransferase remained increased (1301 IU/l) despite good levels of tacrolimus (12 μg/l, treatment range: 5–15 μg/l). Five months after OLT, his fits became more troublesome and he was changed to chloral hydrate, his tacrolimus dose was decreased, immunoglobulin stopped, and prednisolone increased to 16 mg/day. Eight months after OLT, a liver biopsy specimen showed cholestasis, giant cells, and severe bridging fibrosis. At one year, his epilepsy remained difficult to control with deranged liver function and severe haemolysis despite prednisolone (15 mg/day). The serum bilirubin rose progressively to 2580 μmol/l with deteriorating renal function and the child died 14 months after OLT.

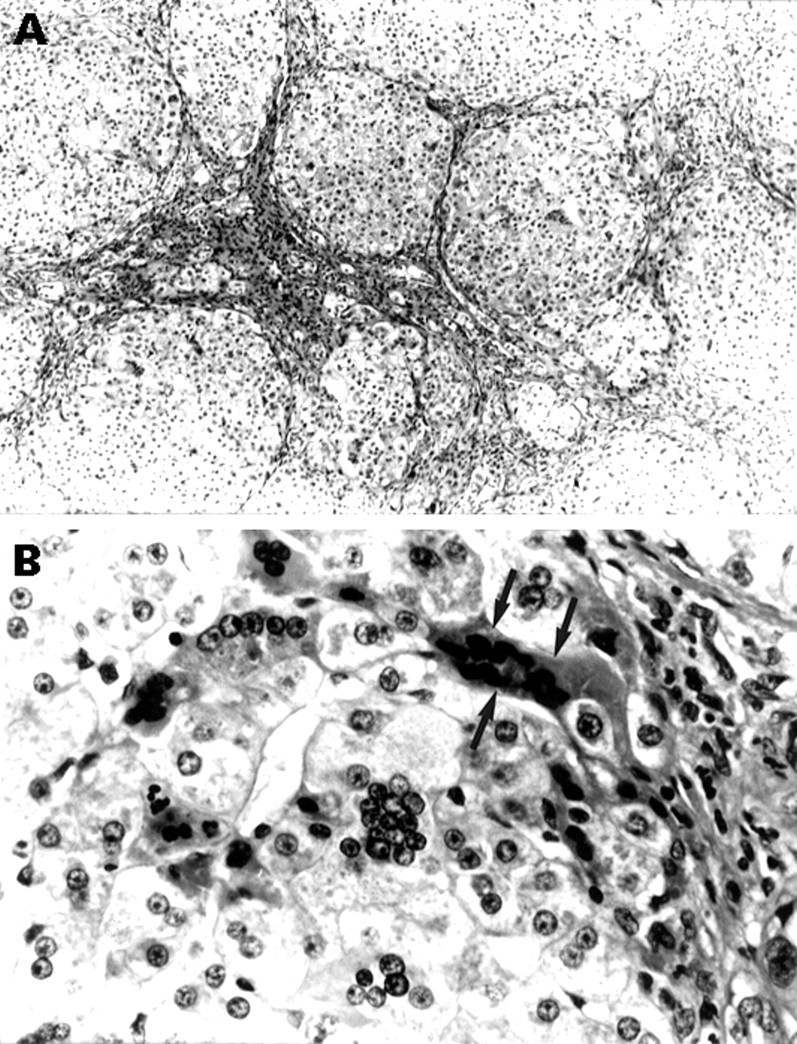{kind=link}
Histopathology of GCH with AHA. (A) Bridging fibrous septa delineate parenchymal micronodules with multinucleated giant hepatocytes (silver for reticulin), and (B) multinucleated giant hepatocytes, one showing eosinophilic shrinkage with nuclear pyknosis (arrows) at the margin of an inflamed fibrous septum (right edge of the field) (haematoxylin and eosin).
Discussion
Giant cell transformation is a common finding in young children with liver disease and an occasional finding in adults. The pathophysiology is not understood, but may represent a non-specific response to liver injury caused by infection, autoimmune disease, cholestasis, drugs, neoplasia, or metabolic disease. Dissolution of intercellular membranes with cellular fusion and nuclear division or cellular growth with nuclear fragmentation secondary to mitotic inhibition have been proposed as possible mechanisms. The potential function of these cells has not been established, but they preserve phagocytic and enzymatic activity and can form and release cytokines. Giant cell transformation has been associated with a variety of liver diseases, including neonatal hepatitis, dysgammaglobulinaemia, HIV, ductal cholestasis, and metabolic disease. In adults giant cell transformation has been associated with autoimmune hepatitis, postinfantile GCH, or syncytial GCH, both reported associated with paramyxovirus, drugs, and hepatocellular carcinoma.
GCH with AHA is a specific entity of unknown aetiology,1although an autoimmune component has been suggested because of the association with Coombs positive haemolytic anaemia and response to immunosuppression. Serological studies for tissue autoantibodies have been negative and the liver histopathology do not meet criteria for diagnosis of autoimmune hepatitis.1-3 Although certain viruses can induce cell fusion in culture, none have been identified in this disorder.1
Clinical features include fever, jaundice, hepatosplenomegaly, and recurrent bacterial infection. Anaemia usually precedes the development of hepatitis.2 Epilepsy is a frequent and severe problem.2 3 The histological appearance is characterised by giant cell formation with intense lobular fibrosis, disruption of lobular architecture, parenchymal collapse, and an overall mild portal inflammatory cell infiltration.1 2 The positive direct Coombs test is intravenous immunoglobulin + C type,1 and liver disease progresses rapidly to cirrhosis and hepatic failure.1 2
Immunosuppression using steroids and azathioprine may produce improvement in the early stages,1-3 however, relapses are frequent and the prognosis appears to be poor. Vincristine, interferon alfa (IFN-α), and intravenous immunoglobulin have been used with partial, or short, lasting remissions. Plasma exchange has been reported to give temporary improvement in severe episodes of AHA, possibly by removing antibodies and toxic substances.
There is little information of the outcome of GCH with AHA after OLT. It was possible to identify three published cases, however, they are seldom recognised as distinct from other types of GCH.4 5All three children (table 1) presented with AHA and developed severe recurrence of GCH with AHA within four weeks of transplant. One child (patient 2) underwent splenectomy and received IFN-α treatment with some improvement, but died from Epstein-Barr virus related lymphoproliferative disease. Another child (patient 3) had acute cellular rejection despite increased immunosuppression and her liver function tests remained abnormal. She was considered for retransplantation, but her parents refused consent, however she was alive at a follow up of six years.
Liver transplantation for GCH with AHA
The association of liver disease and AHA should lead to early liver biopsy to diagnosis GCT, which must be differentiated from other diseases with a different natural history. Immunosuppression may be of value in the early stage of the disease. We found that azathioprine and steroids appeared to be more effective than cyclosporin A, tacrolimus, or immunoglobulin. We did not try IFN-α, and treatment with aciclovir was unhelpful. Liver transplantation in GCH with AHA appears to be associated with a poor prognosis in contrast with other types of GCH and must be considered as a distinct entity.