Article Text

Abstract
The UK Cystic Fibrosis Survey holds data on all people resident in the UK who were diagnosed as having cystic fibrosis and born either since 1968 or before 1968 and alive in 1977. Thus, incidence may be reported from 1968 and prevalence from 1977.The previous estimates are updated to the end of 1995 from data held in the database on 23 August 1996.
The incidence is now calculated as one in 2415 live births. The 1992 mid-year population was 6500 people with 65% aged under 16 years. Births outnumber deaths by 160 per year, which suggests a population of 7750 by the year 2000, with all the increase being in the adult age range.
The survival of successive cohorts continues to be better than earlier cohorts, the linear descent of the curves is still evident. The infant mortality rate for cystic fibrosis is now under 20 per thousand per year and early childhood mortality is under five per thousand per year.
The crude mortality rate for 1995 was 21 per thousand per year, but the standardised mortality ratio was about 3300.
- cystic fibrosis
- epidemiology
Statistics from Altmetric.com
The UK Cystic Fibrosis Survey (UKCFS) has described the basic epidemiology of cystic fibrosis in the UK from 1968 to 1988.1 The UKCFS is one of four total population registers of people with cystic fibrosis, the others being in the Netherlands,2 Sweden,3 and Czechoslovakia.4
The data from the survey are widely used in health service planning, research on cystic fibrosis and in placing the UK experience within an international context.5
This paper updates estimates1 and adds additional years of data up to 1995. All results are presented in a manner consistent with the previous reports.
Methods and analysis
The data collection methods and analysis are effectively the same as in the previous report.1 Three additional surveys have taken place, at the end of 1990 and 1992 and mid-year 1995. Some 1700 clinicians have contributed to the UKCFS; all death certificates for UK residents since 1968 and up to the end of 1995, on which cystic fibrosis or any of its synonyms were mentioned, have been obtained. The UKCFS now holds data on all people who have been resident in the UK with a diagnosis of cystic fibrosis and were born either since 1968 or before 1968 and still alive in 1977. All patients who were alive at any time since 1977 had their diagnosis confirmed by a named consultant. Those who were born and died between 1968 and 1977 were reported by the death certification authorities only. Cases who died before 1968 would have had their cause of death coded using the International Classification of Diseases, eighth revision, which would not always have properly distinguished cystic fibrosis from other conditions. Many patients enjoy shared care between local and regional or national clinics, and great care has been taken to avoid duplicate reporting. Ascertainment is inevitably incomplete for patients born in recent years, either because they are not yet diagnosed or because of delays inherent in the data collection methods used. Experience has shown that there is a delay of about five years before the population estimates become reliable.
The data analysed were those held in the database on 23 August 1996, and the statistical package SPSS-X was used as before.6For the current life table calculation we employed a purpose written program using the methods described by Bradford Hill.7
The 1992 age/sex specific death rates for England and Wales,8 which are very close to the UK rates given the relative sizes of the population, were applied to the 1992 UK population given in table 2 to calculate a standardised mortality ratio (SMR). The number of those aged under 1 year were adjusted to that expected from table 1 in the calculation of the SMR.
Mid-year UK cystic fibrosis population by year, age, and sex based on data held to 1995
Year of birth for UK cystic fibrosis cases, incidence, and estimated births based on data held up to 1995
Results
Table 1 gives the number of people with cystic fibrosis born in the UK per year and the calculated incidence. The average proportion of cystic fibrosis births between 1968 and 1987 is 1/2415, slightly greater than the 1/2475 reported previously.
Table 2 gives the new recorded mid-year UK cystic fibrosis population from 1986 to 1988 and adds the years to 1995. The age group 25+ years has been split into 25 to 34 years and 35+ years except for 1995 which has a 35 to 45 years and 45+ years groups.
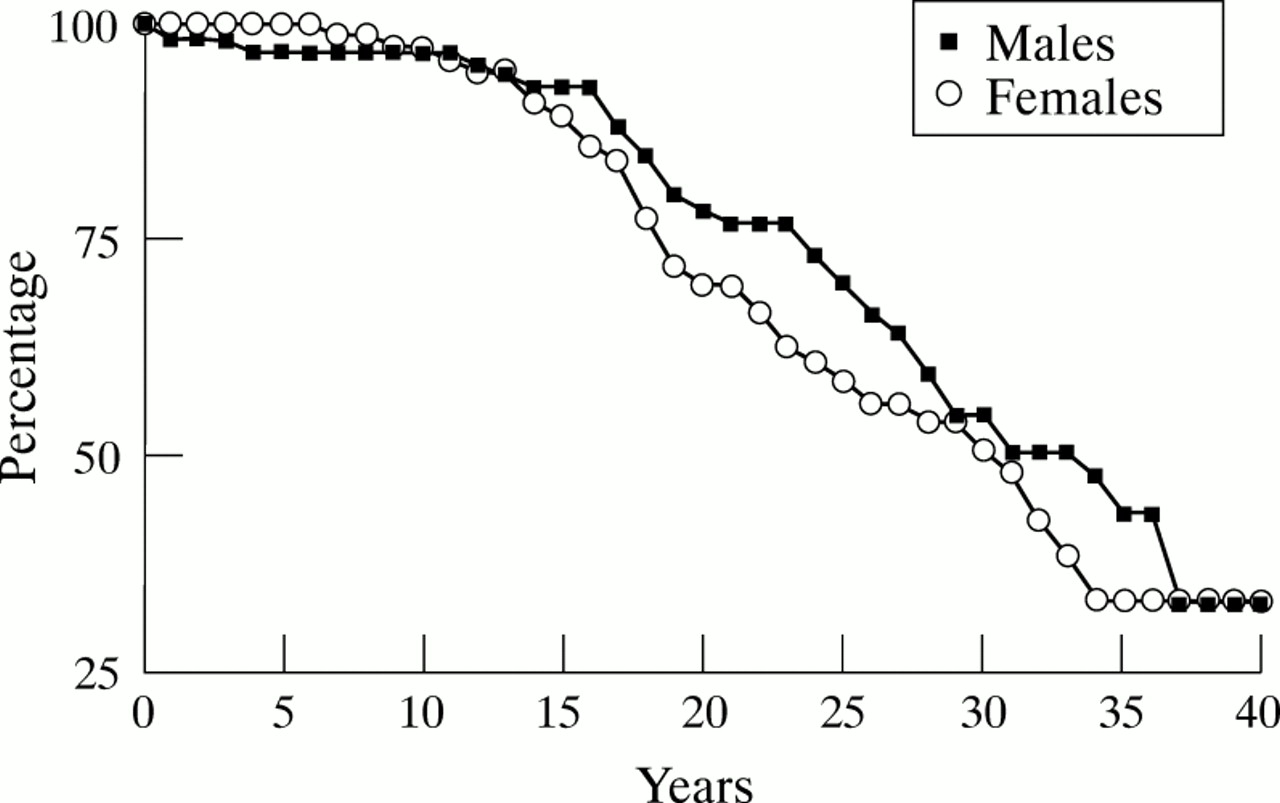
The survival of three year cohorts for the two sexes is given in fig 1with the corresponding hazard rates in table 3. The last two years of each cohort contain censored data and are therefore less reliable than the earlier parts of the curve. The ‘current survival’, based on hazard rates for 1994 is given in fig 2.

Proportion of males and females surviving of each successive three year cohort of UK residents with cystic fibrosis.
Cohort survival of UK cystic fibrosis population: number in population, and hazard rate per thousand by sex (number in parentheses) based on data held to 1995

{kind=link}
{kind=link}
Current survival of UK residents with cystic fibrosis for 1994 for both sexes.
The number of patients dying each year in the decade up to the end of 1995 averaged 138, range 115 to 164. This is some 160 less than the numbers born per year.
Our extrapolation of the population size for the year 2000 is the 1990 population (6244) plus 1500 giving 7750.
The expected deaths in 1992 were 4.4, while the actual deaths were 144, giving an approximate SMR of 3300.
Discussion
The method used to collect data for the UKCFS adds an additional delay to the delays in diagnosis. Experience shows that reliable population estimates for the younger ages only become available after a delay of five years. However, some more recent data are given here to help answer health service planning problems.
The slight increase in the average incidence over the period 1968 to 1985 is due to both an original under ascertainment in the later years, due to delays in diagnosis, together with small numbers of cases being diagnosed in later adult life. For planning purposes the incidence may be assumed as constant at 1/2500, which implies a gene frequency of 1:25.
The 1992 population is already recorded as 6499 people, showing that a previous extrapolation,1 which predicted 6000, was conservative.
Paediatric practice in the UK traditionally continues to age 16; therefore the usual World Health Organisation age groups have been adapted to show the numbers of patients who were eligible for transfer from paediatric to adult care.
The pattern of linear descent of the survival curves continues unbroken after the addition of data up to 1995. The mortality rates for children with cystic fibrosis have now been reduced to such a low level that the prime determinant of the size of the child cystic fibrosis population is the number born. Given the projected cystic fibrosis births based on total UK births and the most recent incidence estimates (table 1), the child population has stabilised at around 4500 cases. All future increase in the population will be in adults and is currently 2600 increasing by about 150 per year.
The final size of the adult population is a matter of conjecture, there being no reliable mortality rates for those aged over 30. We assume that, as is the case for children, adult mortality rates are much lower than they were in 1968–70. Consequently, estimates of continued improvement in median survival9 are now beginning to look realistic. Of course, there is no experience of how cystic fibrosis interacts with the normal aging process, but clinical experience indicates that potentially life shortening complications of cystic fibrosis such as diabetes mellitus and liver disease may become more frequent with advancing age, so projections based on our previous experience of younger people could be erroneous.
Nevertheless, the survival of each successive cohort continues to be more favourable than the previous cohorts. Improvements in mortality may still be possible. However, when compared with the cohorts born since 1983 the margin for improvement is now relatively small and will be difficult to detect given the cohort sizes.
There is still no evidence of a particular age being a crisis point at which the mortality rises sharply. A comparison of the sexes illustrates that, except for the first year of life, the mortality of females is generally greater than that of males. This is consistent with reports from other countries.10
There is no indication that prenatal diagnosis, which has been available in some form for a decade, has had any impact on numbers of cystic fibrosis births but this is not unexpected as most cases are born into families without a history of cystic fibrosis. However, if we have overestimated the number of births since 1991, and if there is any substantial trend towards decreasing incidence, this should become apparent within the next five years.
The numbers dying each year have remained constant at some 160 fewer than those born. This is consistent with improving survival, and the first evidence that improvements in survival are diminishing will be an increase in deaths. As deaths are reported to us promptly by the death certification authorities a worsening of survival would become evident within a couple of years of it occurring. The crude death rate for people with cystic fibrosis has fallen from 40 per thousand per year in 1977 to 21 per thousand per year in 1995. This apparently compares well with the 10.9 per thousand for the general population. However, an SMR for people with cystic fibrosis in 1995 is about 3300, due to the preponderance of young people in the cystic fibrosis population.
To date the UKCFS has found no important systematic differences in either the incidence of cystic fibrosis or the survival of people with cystic fibrosis by region of residence.
These basic epidemiological data provide evidence which should help ensure that planning health care for people with cystic fibrosis is equitable, by reassuring health care purchasers that the levels of provision are not too high and by reassuring patients, their families, and carers that the level of provision is not too low.
Acknowledgments
The UKCFS only succeeds due to the unstinting efforts of the 1700 clinicians and their staff who provide regular updates on their patients.
Mrs Morison is funded by a grant from the UK Cystic Fibrosis Trust.