Article Text

Abstract
OBJECTIVES Despite improved nutrition and intensive treatment, subjects with cystic fibrosis have difficulty in maintaining anabolism during intercurrent infections, which can result in reduced body mass index and impaired skeletal growth. Insulin-like growth factor-I (IGF-I) and its binding protein IGFBP3 are sensitive to changes in nutritional status. The aim of this study was to determine the relation between circulating concentrations of these peptides, body mass index, and clinical status in cystic fibrosis.
METHODS Serum concentrations of IGF-I and IGFBP3 were measured in 197 subjects (108 males, 89 females; mean age 9.69 years, range 0.41-17.9 years) and these data were analysed with respect to body mass index, pubertal stage, and clinical status as assessed by Shwachman score and forced expiratory volume in one second (FEV1 ).
RESULTS The mean height SD score of the children studied was −0.2 (SD 1.14) and the body mass index SD score −0.26 (1.4). The body mass index SD score declined with increasing age (r=−0.18) and paralleled changes in IGF-I concentrations, which also declined. The IGF-I SD score (calculated from control data) correlated with age (r=−0.53). The abnormalities were most obvious during late puberty, when IGF-I and IGFBP3 concentrations were significantly reduced compared with those in control subjects matched for pubertal stage. The IGF-I SD score correlated with height SD score (r=0.14) and the decline in IGF-I concentrations with the fall in body mass index SD score (r=0.42). IGF-I SD scores also correlated with the Shwachman score (r=0.33) and FEV1(r=0.17).
CONCLUSIONS The close relation between declining IGF-I and IGFBP3 concentrations and body mass index in patients with cystic fibrosis may simply reflect poor nutritional status and insulin hyposecretion. Nevertheless, IGF-I deficiency could also contribute towards the catabolism observed in these patients, and IGF-I SD scores correlated with other measures of clinical status such as the Shwachman score and FEV1.
Key messages • The fall in body mass index with increasing age in children with cystic fibrosis parallels the decline in concentrations of IGF-I and its principal binding protein, IGFB3
• The close relation between body mass index and IGF-I concentrations in cystic fibrosis may reflect poor nutrition or insulin hyposecretion
• Nevertheless, low IGF-I concentrations may contribute directly to the fall in body mass index with increasing age
- cystic fibrosis
- insulin-like growth factor-I
- insulin-like growth factor binding protein-3
- body mass index
Statistics from Altmetric.com
- cystic fibrosis
- insulin-like growth factor-I
- insulin-like growth factor binding protein-3
- body mass index
Cystic fibrosis is the most common genetically inherited disease in the UK, with an incidence of about one in 2500. Poor growth, short stature, and delayed puberty have been commonly observed as complications of this ultimately fatal condition and in the past were often regarded as inevitable sequelae of a progressive disease,1-3 but as most patients did not survive into adulthood these observations were of little practical importance for clinical management. Over the past few years there have been major advances in the treatment of cystic fibrosis. Consequently, increasing numbers of adolescents are now surviving into young adulthood with a reasonable quality of life.
Malnutrition in children with cystic fibrosis is associated with poor pulmonary function and poor growth. The cause of malnutrition in cystic fibrosis may be a consequence not only of malabsorption but also of inadequate intake and increased energy expenditure due to recurrent infections.4-7 Aggressive nutritional therapy in patients with cystic fibrosis has contributed to improvements in pulmonary function, growth, the overall clinical course, and survival.8-10 However, despite improved nutrition and treatment regimens many patients with cystic fibrosis still have difficulties in maintaining anabolism during intercurrent infections.
Insulin-like growth factor-I (IGF-I) is an important circulating anabolic hormone. The serum concentration of this peptide is largely dependent on growth hormone.11 In addition to its effects on growth and cartilage metabolism, IGF-I, which is structurally related to proinsulin,12 has insulin-like and mitogenic activity on extraskeletal tissues.13 14
The biological actions of IGF-I are primarily mediated through the type I receptor for IGF, but they are also modulated by a family of six binding proteins, known as the insulin-like growth factor binding proteins, which are present in serum and many body tissues. Insulin-like growth factor binding protein-3 (IGFBP3) is particularly important, binding about 90% of the IGF-I present in serum, and is thought to be an important determinant for IGF-I bioactivity.15 Although primarily dependent on growth hormone, the serum concentrations of IGF-I and IGFBP3 are also under long term regulation by nutritional status.
Despite the importance of IGF-I in normal growth and anabolism, there are few data on IGF-I in cystic fibrosis. Results from previous studies on somatomedin and IGF have been obtained in small numbers of patients with cystic fibrosis and have produced conflicting results, both normal and reduced concentrations having been reported.16 17 A more recent study, however, found reduced concentrations of circulating IGF-I in a large Danish population with cystic fibrosis aged 1–44 years compared with age matched controls.18 In patients who were still in the growth period there was a weak but significant correlation between IGF-I SD scores and height SD scores. Confirmation of these observations, particularly during puberty, and more information on their clinical significance are required.
We report the results of a cross sectional study of 197 subjects with cystic fibrosis aged 0.4 to 17.9 years. We determined IGF-I and IGFBP3 serum concentrations by specific radioimmunoassay and evaluated the relations between these concentrations and body mass index, pubertal development, and overall clinical status as assessed by the Shwachman score and forced expiratory volume in one second (FEV1 (% of predicted values)).
Patients and methods
PATIENTS
A total of 197 patients attending cystic fibrosis clinics for their annual or six monthly assessements at the Royal Brompton Hospital, London, and the John Radcliffe Hospital, Oxford, were recruited for this study. Patients who were admitted for severe infections and other known endocrine disorders were excluded. There were 108 males and 89 females with a mean age of 9.69 years and an age range of 0.4 to 17.9 years.
Control data were obtained from 160 normal subjects who were participating in a longitudinal study of growth and endocrine changes during childhood and puberty.19 20 A subset of 139 subjects were used for the analysis of serum IGF-I concentrations during puberty and 89 for the analysis of IGFBP3 concentrations. The distribution of pubertal stage and sex in the two groups is summarised in table 1.
Number of subjects in different stages of puberty
METHODS
Height was measured using a Harpenden Stadiometer and weight by electronic scales. The body mass index was calculated from the formula weight (kg)/height (m2) and SD scores, were calculated for the patients with cystic fibrosis from published national standards.21 The formula used to derive this and other SD scores was: (observed mean − population mean)/population SD.
Pubertal stage was assessed by the method of Tanner in 108 patients with cystic fibrosis who were over 8 years of age.22Genital stage (G1–G5) for boys and breast stage (B1–B5) for girls were used in all the subsequent analyses.
A quantitative clinical assessment of disease activity, the Shwachman score,1 and FEV1 were recorded for each patient with cystic fibrosis at the time of measurement.
A single random blood sample was obtained from all patients and controls. Serum was separated by centrifugation and stored at −20oC until required for assay.
Serum IGF-I concentration was measured by radioimmunoassay after acid-ethanol extraction of its binding proteins using a polyclonal rabbit antiserum (R557A) raised against purified human IGF-I, as described previously.23 The level of detection of this assay is 5 ng/ml; the interassay coefficients of variation were 9%, 4.5%, and 6.25% at analyte concentrations of 654, 231, and 78.4 ng/ml respectively, with an intra-assay coefficient of variation of 4% at 231 ng/ml.
Serum IGFBP3 concentration was measured by a specific radioimmunoassay. Purified recombinant glycosylated IGFBP3 and rabbit antihuman IGFBP3 antibody were kindly supplied by Drs A Sommers and C Maack (Celtrix, Place, CA, USA). Antigen was iodinated by the chloramine-T method and purified on a Sephadex G-25 column. Separation of bound and free antigens was achieved by the addition of donkey antirabbit Sac-Cel made by Wellcome and obtained from IDS (Northumberland, Tyne and Wear). There was no crossreactivity with IGFBP1, IGF-I, or IGF-II. The interassay and intra-assay coefficients of variation were 11.4% and 4.2% respectively at an analyte concentration of 3.5 μg/ml.
STATISTICAL ANALYSIS
Results are given as the mean (SE), or SD if stated. The significance of differences across pubertal stages between the patients and controls and between the sexes was assessed using a one way analysis of variance, the Kruskal-Wallis test. The Mann-Whitney U test was used to find the significant differences between the patients and controls at each pubertal stage.
Correlation coefficients were calculated to assess associations between variables by simple regression analysis, with the probability of significance derived from analysis of variance and the F test; p values <0.05 were considered to be significant.
ETHICS
All aspects of these studies were approved by the local ethics committees, and informed consent was obtained from both the parents and children.
Results
GROWTH DATA
The mean height SD score in the patients with cystic fibrosis was −0.20 (SD 1.14) (range −4.7 to 5.85) and was significantly different from that in the normal controls (p<0.01). There were no significant differences in the height scores between the sexes, although these were significantly lower than those in the controls in girls but not in boys (mean −0.24 (SD 1.18) (p<0.05) and −0.17 (SD 1.11) (NS), respectively). Height SD scores were significantly lower in pubertal subjects (−0.25 (SD 1.2), p<0.02) than in prepubertal subjects (−0.10 (SD 1.01), NS).
The overall mean body mass index for all of the patients with cystic fibrosis was 16.73 (SD 2.25) kg/m2 (range 12.7-25.0). When expressed as an SD score the value was −0.26 (SD 1.4) (range (−4.14 to 3.87) and was significantly different from that in normal controls (p<0.001).
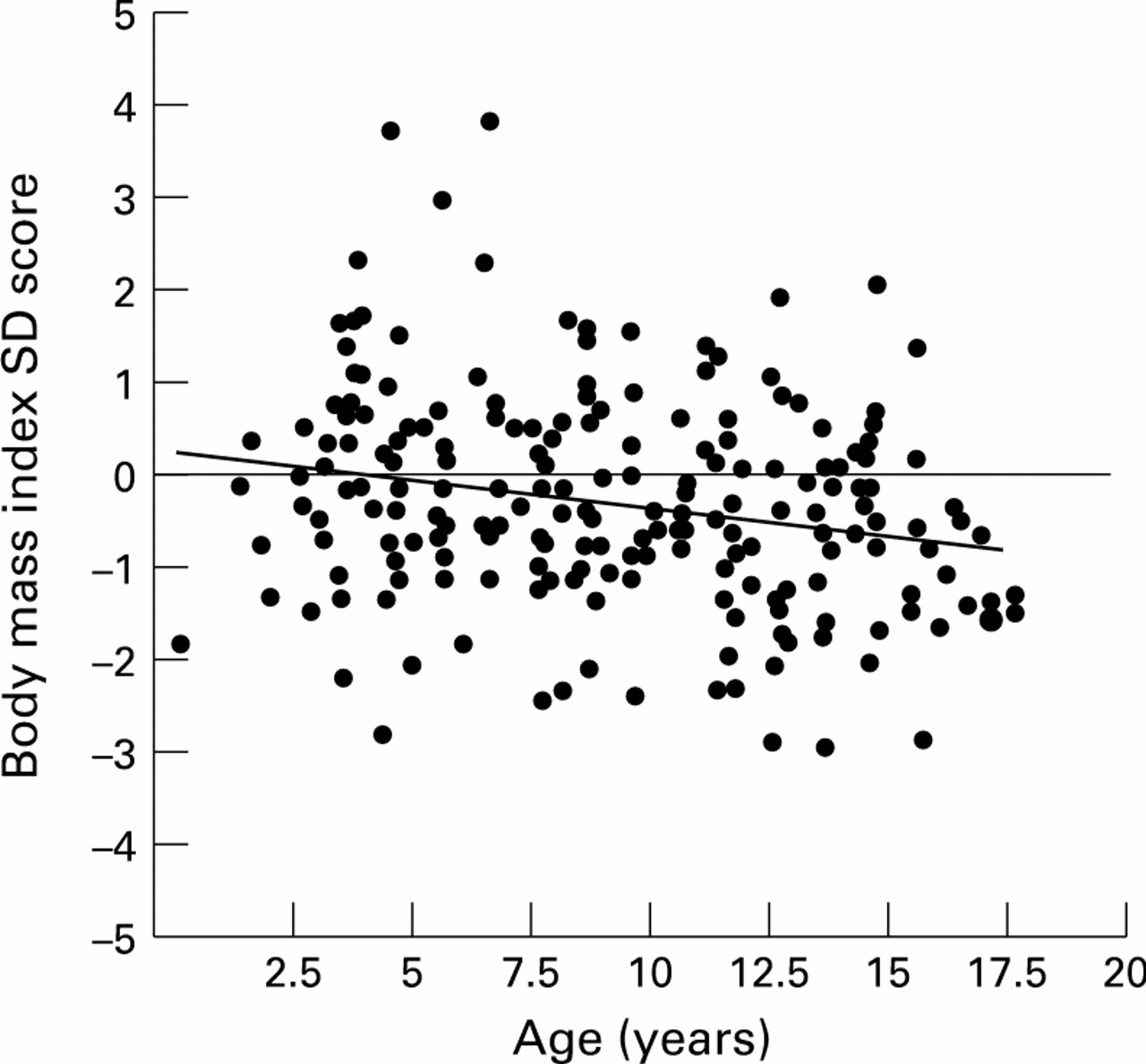
There were no significant differences in body mass index between the sexes in the patients with cystic fibrosis, but body mass index SD scores decreased significantly with increasing age (r=−0.18, p=0.006; fig 1).

Relation between body mass index SD score and age (r=−0.18, p=0.006).
IGF-I AND IGFBP3
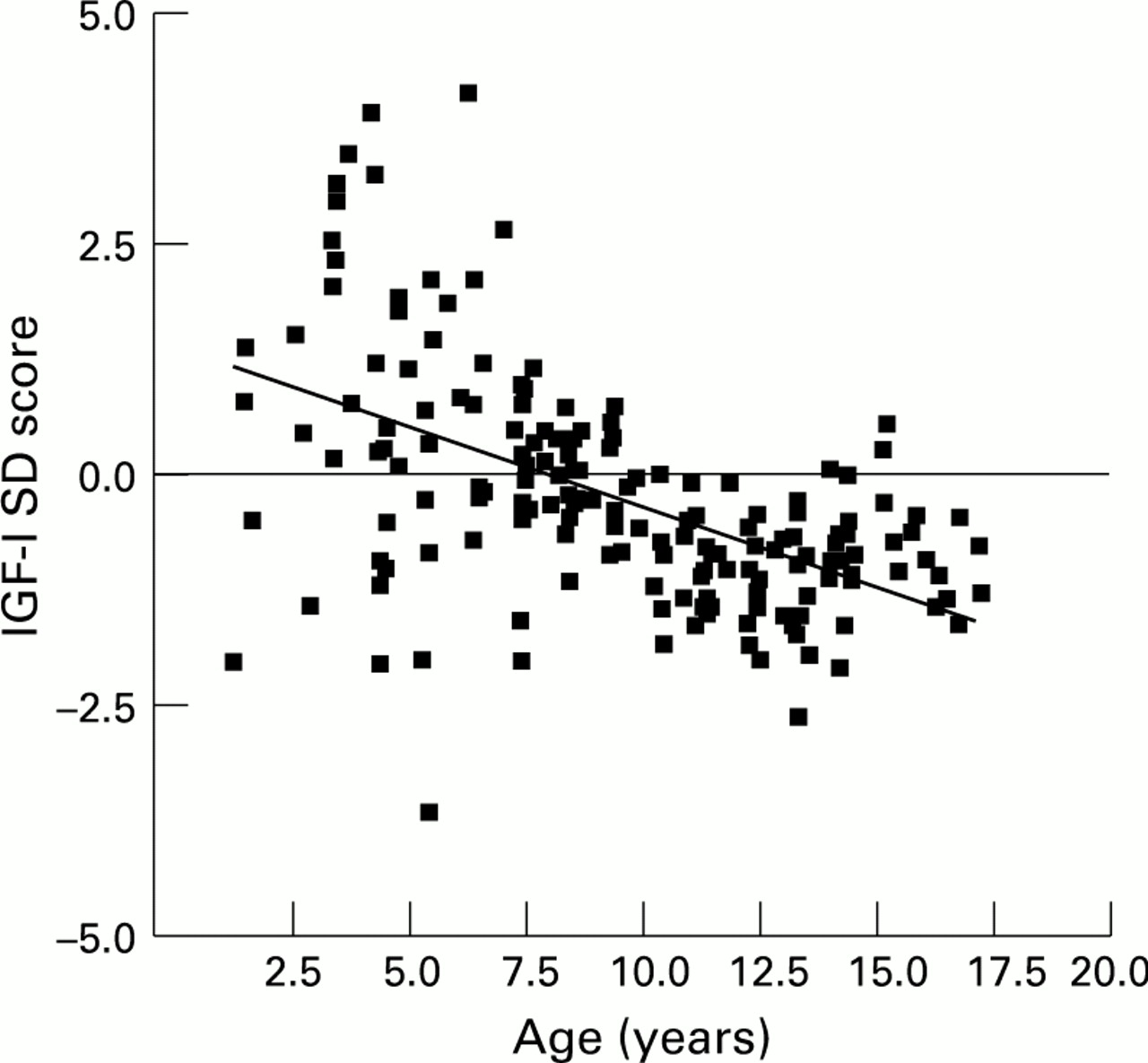
In patients compared with age matched controls serum IGF-I concentrations tended to be low and decreased with increasing age. When expressed as an IGF-I SD score calculated from normal data, the mean IGF-I SD score was −0.14 (SD 1.36) (range −3.62 to 4.46) in the patients with cystic fibrosis. IGF-I SD scores declined with increasing age (r= −0.53, p<0.0001; fig 2). The mean IGF-I SD score in prepubertal subjects was 0.73 (SD 1.75) (range −3.62 to 4.46) and was significantly higher than the mean IGF-I SD score observed in pubertal subjects (−0.61 (SD 0.77) (range −2.55 to 1.21); p<0.0001).

Relation between IGF-I SD score and age in patients with cystic fibrosis (r=−0.53, p<0.0001).
IGFBP3 concentrations correlated with IGF-I values (r=0.42, p<0.0001) and also declined with increasing age compared with controls.
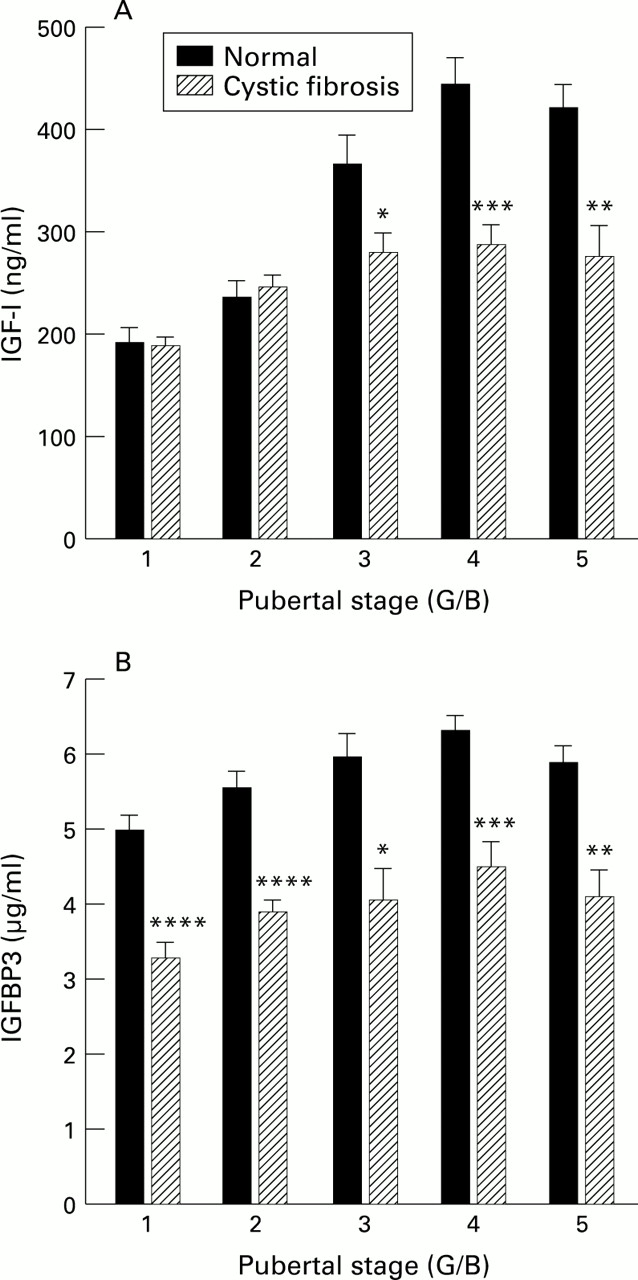
There were no significant differences in IGF-I and IGFBP3 concentrations between the sexes, so data during puberty were pooled. IGF-I concentrations in the patients with cystic fibrosis showed a significant rise during puberty (Kruskal-Wallis test, p<0.0001), but, overall, concentrations were significantly lower than those in the controls through puberty stages G3-G5 and B3-B5 (table 2, fig 3A). IGFBP3 concentrations also significantly increased during puberty (Kruskal-Wallis test, p=0.01) but were significantly lower than those in controls at all pubertal stages (table 3, fig3B).
Effect of puberty on mean (SD) IGF-I concentrations in normal subjects and subjects with cystic fibrosis

(A) Effect of puberty on serum concentrations of IGF-I in normal subjects and subjects with cystic fibrosis (*p=0.04, **p=0.0003, *** p=0.0001). (B) Effect of puberty on serum concentrations of IGFBP3 in normal subjects and subjects with cystic fibrosis (*p=0.002, **p=0.001, ***p=0.0005, ****p=0.0001).
Effect of puberty on mean (SD) IGFBP3 concentrations in normal subjects and subjects with cystic fibrosis
RELATIONS BETWEEN VARIABLES
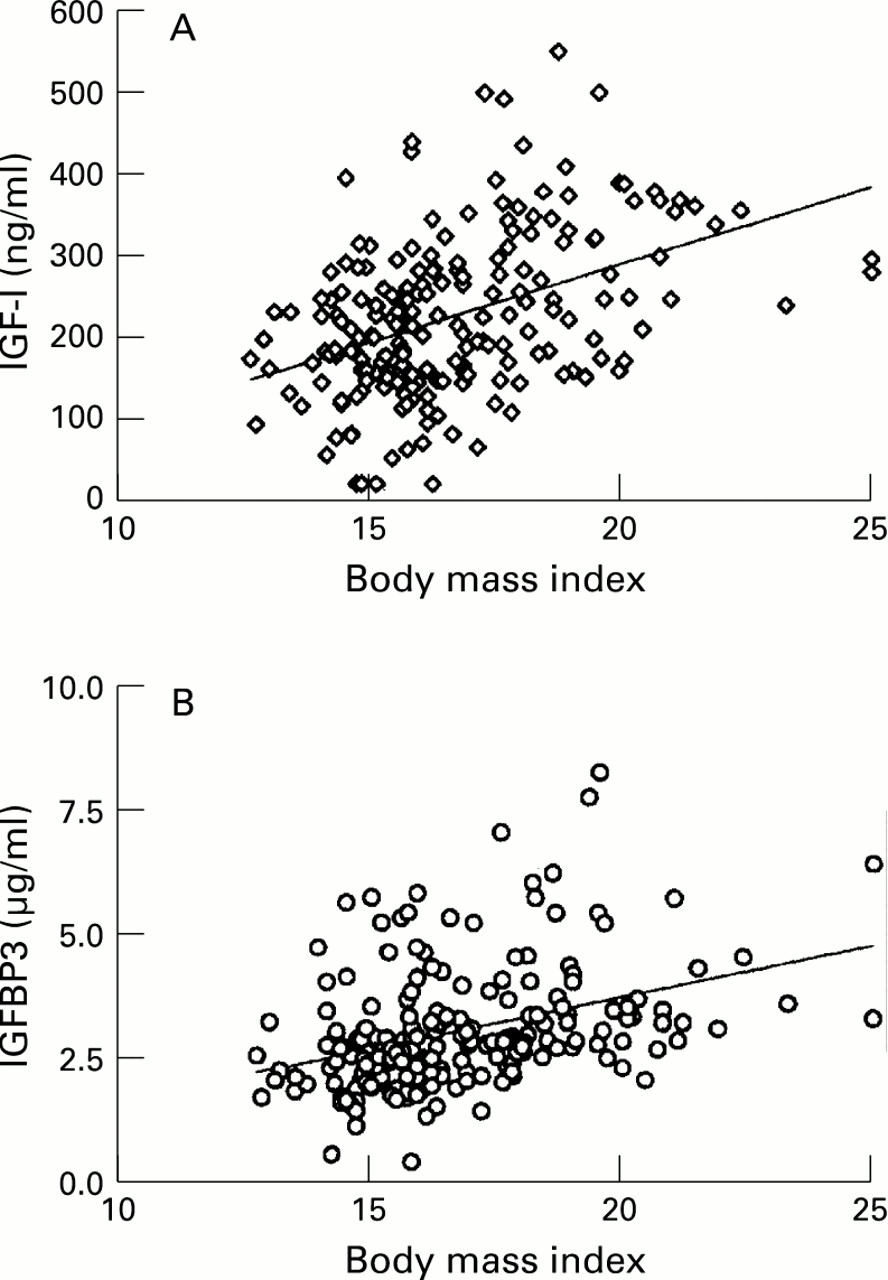
There was a weak but significant correlation between height and IGF-I SD scores (r=0.14, p=0.04). Both IGF-I and IGFBP3 concentrations were strongly correlated with body mass index (r=0.42 (p<0.0001) and r=0.37 (p<0.0001) respectively; fig 4), and there was a significant correlation between IGF-I and BMI SD scores (r=0.16, p<0.02; fig 5)

(A) Relation between body mass index and serum IGF-I concentration in patients with cystic fibrosis (r=0.42, p<0.0001). (B) Relation between body mass index and serum IGFBP3 concentrations in subjects with cystic fibrosis (r=0.37, p<0.0001).
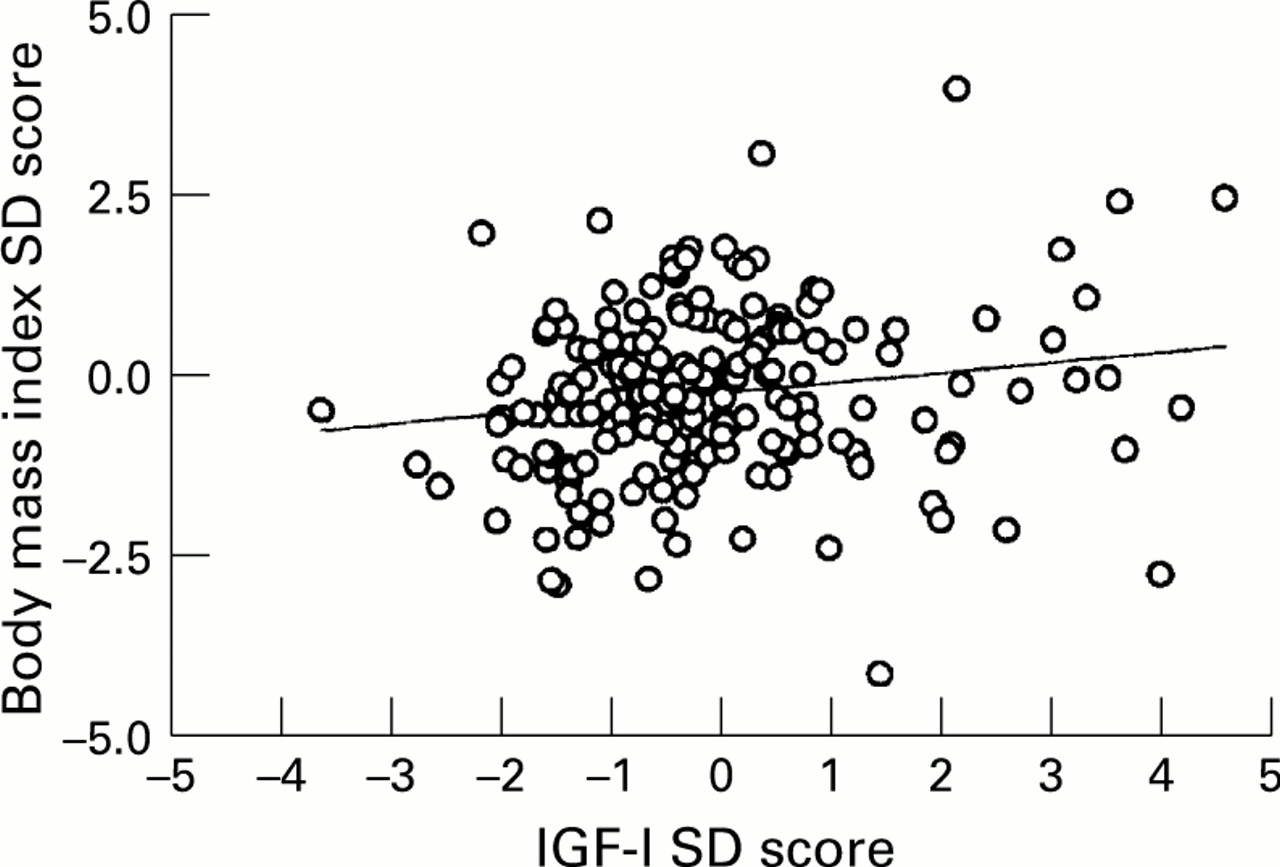
Relation between body mass index SD score and IGF-I SD score in subjects with cystic fibrosis (r=0.16, p=0.02).
IGF-I SD score also correlated significantly with the Shwachman score (r=0.33, p<0.001) and with FEV1(r = 0.17, p<0.02) (fig6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Relation between IGF-I SD score and Shwachman score in subjects with cystic fibrosis (r=0.33, p<0.001). (B) Relation between IGF-I SD score and FEV1 in subjects with cystic fibrosis (r=0.17, p=0.02).
Discussion
These studies confirm the observation made by Laursen et al that IGF-I concentrations tend to be uniformly low in patients with cystic fibrosis.18 These abnormalities are strongly related to age and are most obvious during the latter stages of puberty. As might have been predicted by the binding kinetics, concentrations of IGFBP3 could be correlated with those of IGF-I and, again, concentrations tended to decline with increasing age. The abnormality in IGFBP3 concentration was, if anything, more noticeable than that in IGF-I concentration during puberty.
The changes in IGF-I and IGFBP3 concentrations with age closely paralleled the observed fall in body mass index, and there is a strong correlation between the concentrations of both peptides and body mass index. Like Laursen et al,18 we also found a weak correlation with height SD score. The correlation between height and IGF-I concentrations is similar to that observed in normal subjects, whereas the strong relation between IGF-I and body mass index is not usually evident within the normal range.24 Low IGF-I concentrations are, however, observed in severe malnutrition25-27 and in anorexia nervosa.28Thus malnutrition is at least one explanation for the low IGF-I concentrations in cystic fibrosis.
As well as poor nutrition, recurrent infection, impaired liver function, and insulin deficiency may all contribute not only to reduced body mass index but also to the abnormal IGF-I concentrations in cystic fibrosis. Reduced IGF-I concentrations have been observed in cystic fibrosis during the development of liver impairment,29 but Laursen et al were not able to identify any relation between liver function test results and IGF-I SD scores in their study group.18 There was no clinical or biochemical evidence of impaired liver function in our subjects.
Recurrent infections can clearly have catabolic consequences, resulting in increased protein breakdown and reduced protein synthesis. These effects may be mediated by cytokines such as tumour necrosis factor. The cytokines may have direct catabolic actions, but they may also interact with the IGF axis. The IGF axis in the circulation is disturbed in catabolic conditions,30 31 and cytokines such as tumour necrosis factor can affect IGFBP production and IGF actions at the cellular level.32
Insulin deficiency may be an important determinant of the abnormalities of the IGF-I axis in cystic fibrosis, particularly with advancing age.33 Insulin has an important role in the hepatic regulation of the growth hormone receptor and thus IGF-I production.34-36 Studies of the growth hormone binding protein, which seems to be identical with the extracellular domain of the growth hormone receptor, indicate that this peptide is closely related to both nutritional37-39 and insulin status.40
Reduced concentrations of IGF-I and IGFBP3 are also observed during puberty in children with poorly controlled diabetes mellitus.41 Pubertal growth also tends to be more noticeably affected in pubertal girls than in pubertal boys with diabetes,42 and, interestingly, height SD scores were more reduced in the girls than in the boys with cystic fibrosis in our study. However, in diabetes, in which nutrition is adequate, body mass index is, if anything, increased,43 which is the opposite to what happens in cystic fibrosis. This reflects the relative contributions of insulin and IGF-I in the regulation of growth and weight gain.
Although the regulation of IGF-I production is clearly related to insulin, the distribution of IGF-I and insulin receptors dictate that IGF-I has a greater role than insulin in the regulation of protein metabolism.44 In animal studies IGF-I protects against the catabolic effects of starvation.45 The IGF binding proteins play a critical part in regulating the bioavailability and potential bioactivity of the IGFs,15 and the very low IGFBP3 concentrations that we observed, particularly during puberty, may be important in this respect. In other catabolic states low IGFBP3 concentrations are associated with altered binding affinity brought about by circulating proteases.30 45 This may have the effect of enhancing tissue IGF-I uptake and thus have a protective role by promoting anabolism. Thus the reduced IGF-I concentrations in cystic fibrosis may not only reflect but also contribute to the overall catabolic state, and this is supported by the weak but positive relation we observed between IGF-I and body mass index SD scores in these subjects.
Weight gain is of enormous importance to the overall prognosis in cystic fibrosis as body mass index correlates inversely with overall survival,46 and nutritional status influences the course of the disease.47 It is interesting in this respect that we observed significant correlations not only between IGF-I status and body mass index but also with the more general Shwachman score and FEV1. The effect of improved nutritional intervention and chemotherapy in cystic fibrosis has led to improvements in growth, particularly during adolescence, but the impact on weight for height has been less impressive.4 The persisting abnormalities in the IGF-I and IGFBP axis combined with evolving insulin deficiency may be critical to these observations and warrants further investigation.
Acknowledgments
This study was presented in part at the ninth annual North American cystic fibrosis conference in Dallas, Texas, in October 1995. An abstract is published in Pediatric Pulmonology1995 suppl 12:292 (abstract 293).
We thank the Cystic Fibrosis Trust for its financial support of this study.