
Summary
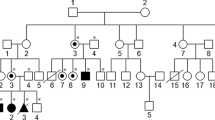
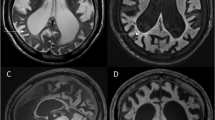
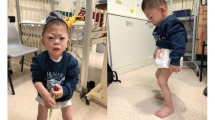
Two patients with a complete deletion of the iduronate-2-sulphatase (IDS) gene are described. In both patients, the resulting phenotype was that of very severe Hunter syndrome (mucopolysaccharidosis II). In addition, both had features not commonly seen in this disorder, e.g. early onset of seizures in one patient and ptosis in the other. It is speculated that loss of adjacent loci may contribute to the unusual findings and that the severe features present in both patients may represent contiguous gene syndromes. Further analysis of IDS cDNA from other patients with Hunter's syndrome may eventually enable phenotype to be predicted more accurately.
Similar content being viewed by others
References
Hopwood JJ (1979) α-l-iduronidase, β-d-glucuronidase and 2- sulfo-l-iduronate 2-sulfatase preparation and characterisation of radioactive substrates from heparin. Carbohydr Res 69:203–216
Hopwood JJ, Harrison JR (1982) High resolution electrophoresis of urinary glycosaminoglycans. Anal Biochem 119:120–127
Hopwood JJ, Morris CP (1990) The mucopolysaccharidoses: diagnosis, molecular genetics and treatment. Mol Biol Med 7:381–404
Lim TW, Leder IG, Bach G, Neufeld EF (1974) An assay for iduronate sulfatase (Hunter corrective factor). Carbohydr Res37:103–109
Nelson PV, Carey WF, Morris CP, Pollard AC (1989). Cystic fibrosis: prenatal diagnosis and carrier detection by DNA analysis. Med J Aust 151:126–131
Neufeld EF, Muenzer J (1989) The mucopolysaccharidoses. In: Scriver CR, Beaudet MC, Sly WS, Valle D (eds) The metabolic basis of inherited disease, 6th edn. McGraw-Hill, New York, pp 1565–1587
Robertson DA, Callen DF, Baker EG, Morris CP, Hopwood JJ (1988) Chromosomal localisation of the gene for human glucosamine-6-sulphatase to 12q14. Hum Genet 79:175–179
Schmickel RD (1986) Contiguous gene syndromes: a component of recognizable syndromes. J Pediatr 109:1169–1173
Suthers GK, Oberlé I, Nancarrow J, Mulley JC, Hyland VJ, Wilson PJ, McCure J, Morris CP, Hopwood JJ, Mandel JL, Sutherland GR (1991) Genetic mapping of new RFLPs at Xp27-q28. Genomics 9:37–43
Wilson PJ, Morris CP, Anson DS, Occhiodoro T, Bielicki J, Clements PR, Hopwood JJ (1990) Hunter syndrome: isolation of an iduronate-2-sulphatase cDNA clone and analysis of patient DNA. Proc Natl Acad Sci USA 87:8531–8535
Wilson PJ, Suthers GK, Callen DF, Baker E, Nelson PV, Cooper A, Wraith JE, Sutherland GR, Morris CP, Hopwood JJ (1991) Frequent deletions at Xq28 indicate heterogeneity in Hunter syndrome. Hum Genet (in press)
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Wraith, J.E., Cooper, A., Thornley, M. et al. The clinical phenotype of two patients with a complete deletion of the iduronate-2-sulphatase gene (mucopolysaccharidosis II — Hunter syndrome). Hum Genet 87, 205–206 (1991). https://doi.org/10.1007/BF00204183
Received:
Issue Date:
DOI: https://doi.org/10.1007/BF00204183