Article Text

Abstract
Objectives To describe the clinical presentation and sequelae, including salt-wasting crises of newly-diagnosed congenital adrenal hyperplasia (CAH) in children aged over 1 year in a contemporary population without screening. To appraise the potential benefit of newborn screening for late-presenting CAH.
Design Active national surveillance undertaken in Great Britain prospectively from 2007–2009 through the British Paediatric Surveillance Unit.
Setting England, Wales and Scotland.
Patients Children first presenting aged 1–15 years with clinical features of CAH and elevated 17-hydroxyprogesterone.
Results Fifty-eight children (26 [45%] boys) aged 1–15 years were reported; 50 (86%) had 21-hydroxylase deficiency. Diagnosis was precipitated by secondary sexual characteristics (n=38 [66%]; median age 5.8 [IQR] 4.8, 7.6) years, genital virilisation (8 girls; 3.2 [IQR 1.3, 7.3] years) or an affected sibling (n=8; 10.0 [IQR 7.4, 13.3] years). At least 33 (57%) children had advanced bone age and 13 (30%) were obese (body mass index ≥95th centile). No child had experienced a salt-wasting crisis.
Conclusions In Great Britain, 30 children aged 1–15 years present annually for the first time with CAH. Older children frequently manifest prematurely advanced epiphyseal and pubertal maturation and genital virilisation, which are often irreversible and likely to have long-lasting consequences for adult health and wellbeing. Almost one-third of affected children are obese before commencing steroid therapy. Newborn screening offers the potential to avoid serious clinical manifestations in older children with unrecognised CAH; however, it may also detect some children who would otherwise remain asymptomatic and for whom the benefit from treatment is uncertain.
- Congenital Abnorm
- Endocrinology
- Epidemiology
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
What is already known on this topic
-
Congenital adrenal hyperplasia (CAH) presenting in older children is often considered ‘late-onset’ or ‘non-classical’, but may also be due to delayed recognition of simple-virilising or salt-wasting forms.
-
If CAH remains untreated due to delayed recognition, excess androgen production can stimulate progressive virilisation, premature epiphyseal and sexual maturation and lead to infertility.
-
CAH is a lifelong, chronic condition and reduced quality of life has been demonstrated in affected adults in the UK.
What this study adds
-
Each year in Great Britain, in an unscreened population, around 30 children aged over 1 year are clinically diagnosed with congenital adrenal hyperplasia.
-
Two-thirds of children presented at primary school age with premature secondary sexual characteristics, 57% children had advanced bone age and 25% girls presented with virilisation.
-
Newborn screening could potentially avert the significant late impacts on health and quality of life, in addition to reducing neonatal mortality and adrenal crises.
Introduction
Congenital adrenal hyperplasia (CAH) refers to a group of recessively inherited enzyme deficiencies influencing adrenal steroid production, leading to decreased cortisol and aldosterone and excessive androgens.1 It affects approximately one in 18 000 liveborn infants in the UK, but not all cases are diagnosed within the first year of life.2 Newborn screening for CAH is not currently undertaken in the UK although this is under review by the National Screening Committee.2
Three forms of CAH are often described: classical CAH comprises salt-wasting (SW) and simple virilising (SV) forms, while a late-onset form is frequently termed non-classical (NC). In newborns, SW-CAH can lead to life-threatening crises due to the inability to maintain sodium balance, while SV-CAH often manifests as female genital virilisation. Key presenting features in older children include premature pubarche and accelerated early growth leading to premature epiphyseal maturation and adult short stature.3 ,4 Recently, concern has also been raised about obesity.5 ,6 If untreated, excess androgen production stimulates progressive virilisation, premature pubic hair development, girls may not develop regular menses and infertility is possible in both sexes.1 Research into the psychosocial impact has hitherto primarily concerned the masculinising effects of excess androgen on the female brain;7 however, reduced quality of life has been demonstrated for adults with CAH.8 Although steroid replacement therapy should avert adrenal crises and facilitate normal development, CAH has a lifelong impact on health.9
While the epidemiology of infants is well-described,2 ,10 ,11 fewer reports exist of clinical presentation with CAH in older children3 in whom life-threatening episodes are rare but who nonetheless experience avoidable adverse sequelae. We used data collected through the British Paediatric Surveillance Unit (BPSU), which obtained over a 2-year period, notifications of all new CAH diagnoses in children aged 15 years and under resident in Great Britain. Specifically, we estimated the incidence of late clinical presentation and its associated complications.
Methods
Active surveillance for CAH in children aged 0–15 years in Great Britain was undertaken prospectively from 2007 to 2009 through the BPSU, a national paediatric rare disease surveillance system involving almost all UK consultant paediatricians (n=3000). Every month paediatricians return a postal Orange card, listing approximately 10 conditions under surveillance, stating the number of children diagnosed with each condition in the previous month or indicating ‘no cases to report’. As described in our earlier paper,2 cases of CAH were defined by specified clinical features in association with elevated plasma 17-hydroxyprogesterone (17-OHP). Clinicians reporting a case subsequently completed a detailed questionnaire using each child's hospital case notes. In accordance with BPSU surveillance methodology, clinicians were not asked to arrange additional clinic visits or to perform any additional tests or examinations. Findings from diagnostic investigations, such as 17OHP, urinary steroids, DNA and synacthen stimulation were reported by clinicians against local age-standardised reference norms. As there was considerable variation in the tests performed and reference standards used in local clinical practice, all completed questionnaires were reviewed centrally by an expert panel of clinicians who determined whether a child had CAH or not on the basis of the clinical presentation and investigations reported by local clinicians.
Age-specific incidence of diagnosis was calculated, excluding children notified in the first reporting month to ensure that prevalent cases were not included. Height and weight z-scores were standardised for age and sex against the British 1990 growth reference.12 Analyses were performed using Stata SE V.11 (StataCorp, College Station, Texas, USA).
Ethical approvals were obtained (ref: 07/MRE12/25; PIAG/BPSU 1-05(FT4)/2007).
Results
During 25 months of surveillance, 144 children with new diagnoses of CAH were identified by clinicians working at 53 institutions. The monthly BPSU card return rate was 92%–94% and the questionnaire completion rate for CAH case reports was 95%. As described previously,2 86 children (36 boys) presented within the first year of life (genital virilisation=37; SW crisis=30, affected sibling=12, other=8). Of 60 children presenting after age 1 year, two with precocious puberty were determined not to have CAH after expert panel review. In this paper, we describe 58 children who presented clinically with CAH aged 1–15 years (median age 5.9 [IQR 4.7, 8.4] years; table 1).
Characteristics of children with late-presenting CAH (n=58)
The annual age-specific incidence (risk) of CAH diagnosis between 1 and 15 years of age (based on 52 children notified between 1 September 2007 and 31August 2009) was 0.23 per 100 000 children (Fisher's exact 95% CIs 0.19 to 0.33) and did not differ by sex (data not shown).
Boys comprised 45% of those with late-presenting CAH (n=26; 95% CI 32% to 58%). Median age at first clinical presentation was similar for both sexes (table 1; Mann–Whitney p=0.40). The most frequently encountered subtypes were 21-hydroxylase and 11β-hydroxylase deficiency (table 1). Forty children (69%) were white British and 12 (21%) were Asian/British Asian ethnicity.
Diagnosis was most frequently precipitated by presentation with secondary sexual characteristics (table 1), which occurred at a median age of 5.8 (IQR 4.8, 7.6) years. Four additional children, investigated after diagnosis of CAH in a sibling also demonstrated secondary sexual characteristics. Eight girls first presented with virilisation: six at Prader stage 1and two at stage 3 or 4. Nine girls who presented due to secondary sexual characteristics or an affected sibling were also virilised (Prader 1 or 2). Of eight children investigated only after a sibling was diagnosed, six had clinical signs (table 1). No child experienced adrenal crisis, defined as a life-threatening state of cardiovascular collapse; however, five children manifested adrenal insufficiency (biochemical changes indicating inadequate cortisol production) at first presentation and, in one additional child, this developed subsequently. Median interval from initial suspicion to confirmation of CAH was 33 days (IQR 14, 59 days; n=51) and did not differ by sex or ethnicity (data not shown).
Median 17-OHP concentration reported for 50 (86%) children at presentation without synacthen stimulation was 95 (IQR 25, 208) nmol/L. Subsequent diagnostic investigation varied; 32 children had a synacthen stimulation test, reported as ‘abnormal’ in all cases and 40 children had urinary steroid profiling which was diagnostic in 38 cases. DNA results were available for 16 children (table 1; DNA results unavailable=15). The most frequently observed mutations were g.999T>A (9 children), g.655A/C>G (5 children) and g.1683G>T (4 children).
At notification, 48 (83%) children had commenced glucocorticoid, and seven, mineralocorticoid replacement therapy. Almost 90% (n=52) were referred to another specialist, usually a paediatric endocrinologist (n=42) or geneticist (n=21).
Although 32 children were reported to have ‘accelerated growth’, this precipitated diagnosis in only two; however, routine growth monitoring in England is undertaken only at ages 5 and 11 years.13 Bone age was assessed in 36 (63%) children and was advanced in 33, appropriate in two and retarded in one relative to chronological age (magnitude not reported).
Height, weight and body mass index (BMI) SD scores (SDS) at diagnosis were estimated for 44 children (figure 1). Height SDS was >2.0SD in 39% (17/44) children, all diagnosed aged under 11 years, but <1.0SD in all four children diagnosed after 11 years. Weight SDS was >2.0SD in 41% (18/44) children and did not show any clear pattern with age. BMI ranged from 14 to 37; 13 (30%; 4 boys; 9 girls) were obese (BMI ≥95th centile13).
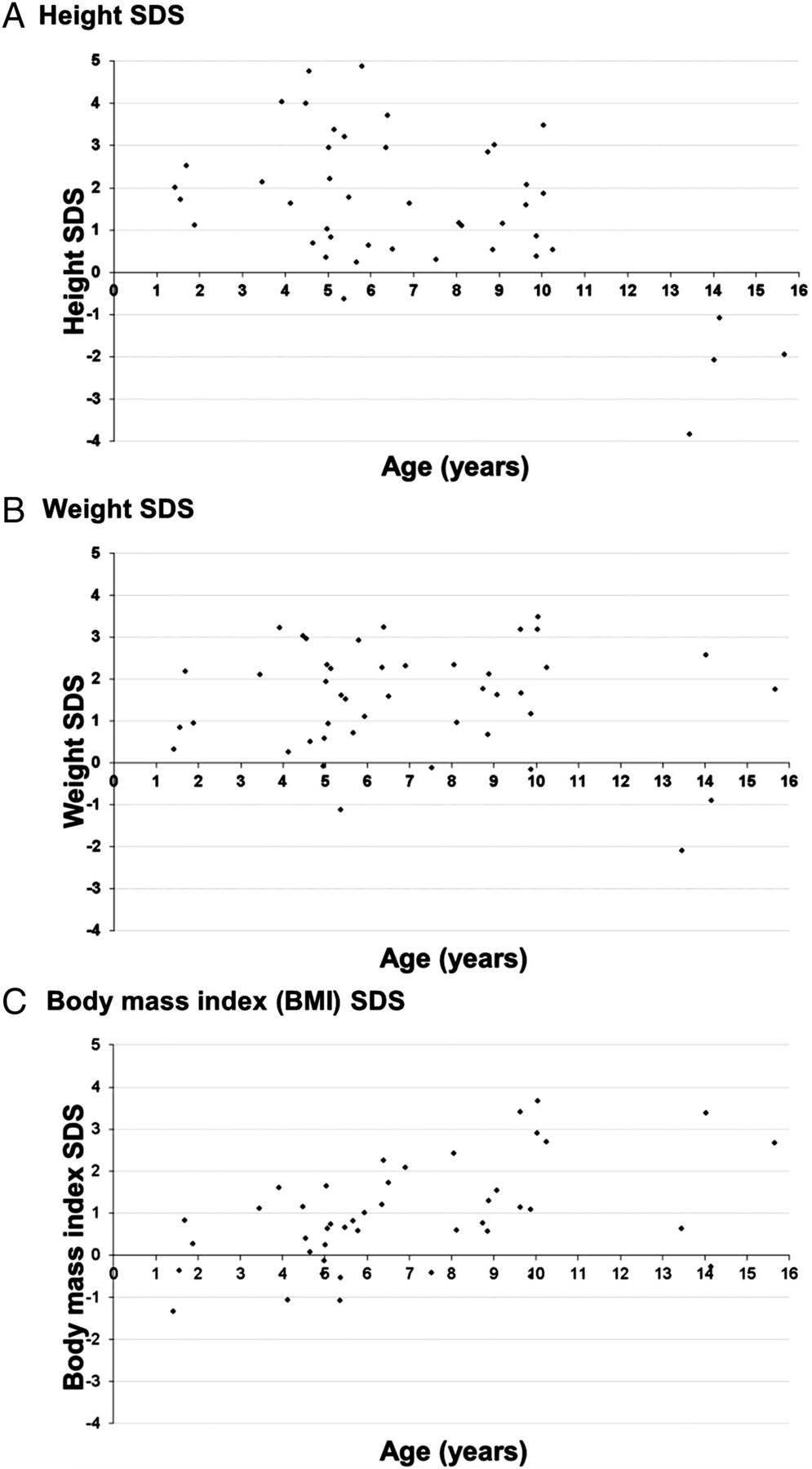
{kind=link}
SD scores (SDS) for height and weight at first presentation with congenital adrenal hyperplasia for children over 1 year of age (n=44). (A) Height SDS. (B) Weight SDS. (C) Body mass index (BMI) SDS.
Discussion
We report the incidence and clinical features of new diagnoses of CAH in children aged over 1 year in Great Britain ascertained through active national surveillance of over 3000 paediatricians with a 94% monthly response rate. We estimate 30 children aged 1–15 years are diagnosed annually with CAH with over-representation of children of Asian ethnic origin.14 The age-specific risk of new CAH diagnosis aged 1–15 years is low at 0.23 per 100 000 in comparison with 5.5 per 100 000 children aged under 1 year,2 suggesting that most affected children are diagnosed during infancy.
Diagnosis in three-quarters of children was precipitated by premature secondary sexual characteristics at a median age of 6 years. At diagnosis, 50% of girls had virilised genitalia and at least half of all children had advanced bone age. Although one in seven children were investigated only after their sibling had been diagnosed, most had signs of CAH when examined. Premature pubarche, genital virilisation and advanced bone age are largely irreversible and have long-lasting implications for health. While the rationale for early detection is primarily prevention of SW crises, newborn screening also offers the potential to avoid serious clinical manifestations of CAH in older children.
Major strengths of this study were the high case ascertainment and questionnaire completion rate due to active surveillance. Nevertheless, it is possible that some affected children were not reported by clinicians or remain undiagnosed, particularly those with mild or asymptomatic CAH, resulting in a bias towards reporting more severe cases. Laboratory surveillance did not have coverage of the whole UK and was not fully independent. So, we were unable to perform capture-recapture15 analyses to estimate completeness of ascertainment. As we were restricted to requesting information available from case notes, some analyses, such as gene mutations were limited by clinical practice and small sample sizes. NC-CAH phenotype has been associated with g.1683G>T and g.89C>T gene mutations, which were the least severe mutations found in six study children. In eight further children in our sample, the least severe mutation was g.999T>A and in two children the mutation was g.655A/C>G; these have been associated with SV-CAH and SW-CAH phenotypes respectively.16 Speiser has suggested that the penetrance of genes for NC-CAH varies between populations.17
Previous population-based paediatric surveillance studies in Australia10 and the Netherlands,11 report little detail of postinfant diagnoses. Thilen's3 retrospective case series from Sweden provide data from an unscreened population during 1969–1986 and included 29 children presenting between 1 and 14 years of age; median age at diagnosis was 4 years and median delay in confirming the diagnosis was 17 months after initial symptoms. Children diagnosed after 1 year old had bone ages advanced by a mean 55 months and final height for most was 1.0SD below the population mean. Eugster4 demonstrated that children treated appropriately for CAH achieved a final height within 1.0SD of the mid-parental prediction and emphasised that diagnosis within the first year of life was a crucial factor. Nevertheless Nordenstrom has cautioned that, with the advent of newborn screening in Sweden, some children with mild disease may be receiving excess steroid replacement therapy and experiencing a detrimental effect on growth.18 Our data from an untreated population of children with CAH are consistent with previous findings of accelerated epiphyseal maturation leading to short stature. Although children diagnosed after age 11 years within our study had reduced height z-scores, we did not collect longitudinal data or parental heights, so cannot draw conclusions about target height or individual growth trajectories.
Glucocorticoid therapy does not provide physiological steroid replacement and an association with obesity, hypertension and metabolic syndrome in children with CAH has been proposed; there is also increasing evidence that CAH-related obesity places affected adults at higher risk of cardiovascular complications.5 ,6 ,9 In our study, we found that 21% boys and 41% girls aged 1–15 years were obese at the time of first diagnosis and prior to commencing steroid replacement therapy; this compares with 10% boys and 9% girls aged 5 years, and 20% boys and 17% girls aged 11 years in England whose BMI ≥95th centile during an equivalent time period.13 Our findings of obesity in an untreated population of children and adolescents with CAH support earlier studies reporting a higher risk of obesity in individuals with CAH.5 ,6 ,9 Previous authors have proposed that the causes of obesity in children and adults with CAH are multifactorial and our study provides additional evidence that the disease process influences weight gain independently of glucocorticoid therapy.
Our prospective population-based study has high ascertainment of clinically diagnosed cases within an unscreened population. Some authors19 have concluded that increased clinical awareness has reduced adverse neonatal outcomes and questioned the need for newborn screening, while Grosse20 has suggested that deaths with undiagnosed CAH are lower than previously estimated. Screening is usually advocated on the basis of averting neonatal crises; measuring the burden of disease in older children, who in our study already had evidence of genital virilisation, secondary sexual characteristics, accelerated growth and/or obesity at diagnosis, is highly relevant to understanding whether newborn screening would have additional benefit in preventing adverse outcomes in children who represent late-onset or perhaps ‘missed’ mild cases of CAH. Screening based on biochemical or genetic markers in the newborn bloodspot should be approached with caution, however, as it might also detect children with CAH who would otherwise remain asymptomatic. For these children, the benefit from treatment is uncertain, particularly in view of the potential negative effects of glucocorticoid therapy highlighted by Nordenstrom.18 Our data suggest that some children with previously unrecognised CAH do seek treatment for clinical manifestations of the condition during later childhood and earlier detection would increase the potential for improving therapy and interventions to optimise their final height and weight. Nevertheless, the impact on mild or asymptomatic CAH should remain a key consideration when determining the implications of introducing a newborn screening programme.
Late childhood diagnosis of CAH has significant implications for adult health and early detection and treatment might prevent adverse clinical progression and irreversible sequelae. Carroll estimated the benefit of newborn screening for CAH at $20 357 per quality adjusted life-year gained.21 The anticipated benefits of newborn screening for CAH should not therefore be confined to preventing mortality and serious neurological morbidity due to neonatal adrenal crises, but should also include the potential to avert any significant late impacts on adult health and quality of life.
Acknowledgments
We acknowledge the BPSU, supported by the Department of Health for facilitating this study. The views expressed in this publication are those of the authors and not necessarily those of the Department of Health or BPSU. We thank Mr Richard Lynn and Ms Helen Hopkins (nee Friend) at the BPSU as well as all reporting clinicians and participating laboratories, for their help with data collection. We are also very grateful to the British Society of Paediatric Endocrinology and Diabetes and ‘Living with CAH’ (formerly Climb-CAH) for supporting this study.
References
Footnotes
-
Collaborators Participating laboratories: Glasgow Royal Infirmary, Glasgow; Addenbrooke's Hospital, Cambridge; University College London Hospitals, London; Belfast Royal Victoria Hospital, Belfast; Southampton General Hospital, Southampton; Barts and the London NHS Trust, London; SAS Steroid Hormone Centre, United Leeds Hospitals, Leeds; St. Thomas’ Hospital, London; St Mary's, Imperial College Healthcare NHS Trust, London; Royal Gwent Hospital, South Wales, Newport, Gwent; Royal Liverpool University Hospital, Liverpool; West Park Hospital, London; Royal Manchester Children's Hospital, Manchester.
-
Contributors We confirm that all coauthors made a substantial contribution to the study design, data analysis and drafting of this manuscript. All authors taken part in the writing and final editing of the report, all have been given a copy of the manuscript, all have approved the final version of the manuscript and all are prepared to share responsibility and accountability for the results. CD initiated the collaborative project, gave input on the study design and on the data collection, reviewed the data and revised the draft paper. RLK implemented the study, gave input on the study design, monitored data collection, analysed the data and drafted and revised the manuscript. JMK implemented the study, gave input on the study design and data collection tools, monitored data collection, performed data analyses and revised the draft paper. JO implemented the study, gave input on the study design, monitored data collection and revised the draft paper. PH and CK gave input on the study design and data collection, reviewed the data and revised the draft paper.
-
Funding The UK Collaborative Study of Newborn Screening for CAH was funded by a grant from the Department of Health (England) and commissioned by the UK National Screening Committee. The Centre for Paediatric Epidemiology and Biostatistics benefits from funding support from the Medical Research Council in its capacity as the MRC Centre of Epidemiology for Child Health [grant reference G0400546]. This work was undertaken at Great Ormond Street Hospital/UCL Institute of Child Health which received a proportion of funding from the Department of Health's NIHR Biomedical Research Centres funding scheme.
-
Competing interests None.
-
Ethics approval Thames Valley Research Ethics Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Data were collected under Section 60 (PIAG) approval and cannot be shared.