Article Text

Abstract
To evaluate and manage epileptic seizures and other paroxysmal events in infants, it is necessary to ask five key questions: (1) Is this a type of epilepsy?; (2) What seizure type(s) are occurring?; (3) Do these seizure types, combined with factors such as age at onset and EEG features, constitute an ‘epilepsy syndrome’?; (4) What investigations do we need to do in searching for an underlying aetiology? and finally, (5) What is the prognosis for neurological and developmental state in later life?
This review considers epilepsies that have an onset in infancy but after the perinatal period, outlines the commoner epilepsy syndromes occurring in this age group and describes paroxysmal events that can mimic epilepsy. Epilepsies in infancy may be the manifestation of a genetic predisposition associated with a benign course and good prognosis for neurodevelopment. In contrast, they may pose the challenging situation of ‘epileptic encephalopathy’, rare but potentially treatable metabolic conditions, or structural abnormalities with poor developmental outlook and intractable seizures. Seizures in infancy are relatively rare and there is a wide range of underlying causes, some of which require specific treatments to avoid preventable neurodevelopmental damage. Guidance from the National Institute for Health and Clinical Excellence suggests early referral of cases of infantile epilepsy to a tertiary centre.
- Epilepsy
- Syndrome
- Infancy
- Seizure
- Encephalopathy
Statistics from Altmetric.com
Introduction
In evaluating seizures and other paroxysmal events in infants, it is useful to ask the following five key questions: (1) Is this a type of epilepsy?; (2) What seizure type(s) are occurring?; (3) Do these seizure types, combined with factors such as age at onset and EEG features, constitute an ‘epilepsy syndrome’?; (4) What investigations do we need to do in searching for an underlying aetiology? and finally, (5) What is the prognosis for neurological and developmental state in later life?
If we consider the first question to encompass a detailed and chronological description of the events (the ‘phenomenology’ or ‘semiology’), and the final question to deal with behaviour, cognition and emotional and social functioning, then we can consider these five questions to have a useful correspondence with the five ‘axes’ proposed as a classification scheme by the International League Against Epilepsy (ILAE) in 2001.1
In this review, we consider epilepsies that have an onset in infancy but exclude seizures occurring in the perinatal and early neonatal period, which is a large subject in its own right. In particular, we outline paroxysmal events that mimic epilepsy and we describe the more common epilepsy syndromes occurring in this age group. And we suggest features that distinguish ‘benign’ from more severe epilepsy syndromes, these latter comprising a group that is often referred to as ‘epileptic encephalopathies’. The focus here is on the recognition of key features rather than detailed management, but such recognition is vital for identifying appropriate interventions and predicting likely prognosis.
Question 1: Are the events likely to have an epileptic or non-epileptic basis?
Even in specialist centres, there are high false-positive rates for the diagnosis of epilepsy. This can result in missing other important diagnoses and exposing infants to unnecessary investigations and ineffective treatment interventions. Table 1 shows some of the commoner non-epileptic paroxysmal events that can mimic infantile epilepsies. We consider them in seven groups, although this is not a formal classification: (1) mainly rapid movements; (2) mainly sustained movements; (3) with eye movements as a prominent features; (4) complex movements and behaviours; (5) episodes with associated colour changes, loss of consciousness or weakness without a cardiac cause or (6) such episodes with a possible cardiac cause and (7) other metabolic, iatrogenic or induced illnesses.
Some conditions that might mimic epilepsy in infancy
In particular, care should be given to identifying potentially life-threatening conditions, such as long-QT syndromes and tetralogy spells, or manifestations of serious conditions like severe gastro-oesophageal reflux (Sandifer syndrome) or neuroblastoma (opsoclonus-myoclonus syndrome). Diagnostic tips are shown in table 1.
Epilepsy should be distinguished from acute symptomatic seizures, which arise as the immediate result of a variety of acute insults, such as trauma, infection, hypoxia-ischaemia, hypoglycaemia, hypocalcaemia or other metabolic derangements. In contrast, epilepsy—perhaps better referred to as ‘epilepsies’ since their features and consequences are so varied—is considered to be unprovoked. (However, some epilepsies can be reflex, such as photic-stimulation-induced or reading epilepsies. In infants, reflex epilepsies are rare, but epileptic seizures can be caused by, for example, bathing in hot water.) If seizures recur when the original provoking factors have been removed, they are referred to as ‘remote symptomatic epilepsies’. An example would be infantile spasms and focal seizures developing in a 6-month-old infant who had had a perinatal stroke.
Febrile seizures are the most common form of seizure in infancy.2 They occupy an ambiguous position but are probably best considered to be a form of acute symptomatic seizure rather than as a form of epilepsy, although this is arguable and there is an element of biological and genetic susceptibility. There is a large geographical variation that may be partly genetic and partly related to variations in exposure to the infectious agents precipitating the fever. On the island of Guam, for example, the reported cumulative incidence of febrile seizures in childhood is 14%, in contrast with Western Europe and North America, where it is <5%.
Simple febrile seizures, as opposed to complex, are generalised (tonic-clonic in majority), short in duration (<15 min) and do not recur within the same febrile illness. The risk of developing epilepsy is six times greater than the normal population (ie, 3%), and the risk of developing epilepsy increases to 50% if all three of the following factors are present:
-
abnormal neurodevelopmental status before the first episode;
-
family history of epilepsy;
-
complex febrile seizures.
It is important to avoid premature diagnostic closure in infants who have had previous febrile seizures since any seizure associated with fever might be due to meningoencephalitis, and febrile seizures can precede genuine epilepsy conditions, such as mesial temporal sclerosis and Dravet syndrome (see table 2). It is important to distinguish simple from complex febrile seizures, and to pay particular attention where febrile status epilepticus and/or prominent focal or lateralising features, either ictally or postictally, are present. It is also worth considering the family history in the context of the genetic/familial epilepsy syndromes such as generalised epilepsy with febrile seizures plus.
Commoner epilepsy syndromes with onset in infancy
Question 2: What seizure type(s) are occurring?
A broad range of seizures occur in infancy. The 2001 ILAE proposal suggests classifying seizures into two main groups—self-limited seizures and continuous seizures, the latter being the various forms of convulsive and non-convulsive status epilepticus.1 In either of these groups, seizures can be generalised or focal, which the proposed scheme describes in detail.
Routine interictal EEG has limited predictive value and should be used with caution as a means of identifying whether paroxysmal episodes are due to epilepsy or another paroxysmal condition. Some epileptiform abnormalities, such as central sharp waves, are common in conditions such as cerebral palsy even in the absence of witnessed seizures, and the absence of epileptiform discharges on an interictal EEG would never preclude epilepsy. On the other hand, EEG can help greatly to categorise seizures—the ‘gold standard’ being ictal video-EEG.
Some key points about seizure classification are:
-
Home video may be invaluable and can be incomparably better than a poorly remembered event described verbally by a parent woken by a nocturnal event.
-
Since even video-EEG can be difficult to interpret, surface electromyography, which shows the duration of muscle contraction, can help to distinguish seizure types. For example, myoclonus (duration up to 200 ms) versus epileptic spasms (duration usually 1–2 s) versus brief tonic seizures (several seconds in duration) versus atonic seizures or ‘negative myoclonus’ (absent surface EMG trace).
-
Generalised seizures might be focal at onset and secondarily generalised, so avoid unjustifiable assumptions and be mindful of subtle focal seizure onset.
-
Treatment choices are often made on the basis of seizure types rather than epilepsy syndromes or aetiology.
-
Classification schemes evolve as insights and debate continue, and some terms are confusing or redundant. For example, the term ‘astatic’ indicates a fall from a standing position, which could be due to an atonic seizure (negative myoclonus), a violent myoclonic seizure or a brief tonic seizure. It is a good generic term for a ‘drop attack’ but a poor term for a seizure type.
Question 3: Does this constitute an ‘epilepsy syndrome’?
An ‘epilepsy syndrome’ indicates a confluence of clinical features: typically a characteristic combination of age at onset, type or combination of seizures, and EEG pattern. The characteristic EEG pattern may be ictal and/or interictal. Identifying an infantile epilepsy syndrome:3
-
helps in the choice of first-line treatment interventions;
-
suggests a range of likely underlying causes;
-
suggests a likely prognosis for seizure control and neurodevelopment;
-
provides one form of inclusion criterion for clinical studies.
However, at least a quarter of the epilepsies that occur in infants do not easily fit into ILAE epilepsy syndrome classifications, and there is always a degree of inter-rater disagreement.4 Classification remains a dynamic process with the relationships between infantile epilepsy syndromes and aetiology being refined using video-EEG, improved brain imaging and new diagnostic techniques. Even seizure classification in infants can be particularly challenging.5
Table 2 outlines a number of epilepsy syndromes in infancy and their crude distinction into two categories: those with good prognosis and those who are likely to be challenging to treat.
Challenging epilepsy syndromes: epileptic encephalopathies
The concept of the ‘epileptic encephalopathy’ is that ‘the epileptic activity itself may contribute to severe cognitive and behavioural impairment above and beyond what might be expected from the underlying pathology alone (eg, cortical malformation), and that this can worsen over time’.12 It is regarded as a ‘pragmatic grouping’ rather than a formal element of classification. The best-known examples are Ohtahara syndrome, West syndrome, and Lennox–Gastaut syndrome, but early myoclonic encephalopathy and Dravet syndrome are also generally considered to have the features of an epileptic encephalopathy. An implication is that the current and future cognitive and behavioural state can be modified by effective treatment aimed at controlling the epilepsy. This concept is supported by two retrospective studies of infantile spasms, where shorter lead times between symptom onset and treatment were shown to be associated with better long-term neurodevelopment.13 ,14
Question 4: Can we identify an underlying aetiology?
Symptomatic epilepsy syndromes occur as a consequence of an identifiable primary cause such as a structural, neurodegenerative, metabolic, genetic or chromosomal disorder. An increasing number of genetic causes are being identified.15 West syndrome, the commonest epilepsy syndrome in infancy, is a good example of how epilepsy syndromes may have a variety of possible causes. Since optimal treatment and prognosis are strongly influenced by aetiology, it is best to consider this as a heterogeneous group of conditions sharing a non-specific phenotype: a series of infantile spasms. The diagnostic investigations suggested in figure 1 are adapted from a US expert consensus opinion as to how a clinician is recommended to approach an aetiological diagnosis.16 Although most cases of infantile spasms have identified underlying causes (and are referred to as having a ‘symptomatic aetiology’), about 30% are categorised as ‘cryptogenic’ or ‘idiopathic’.

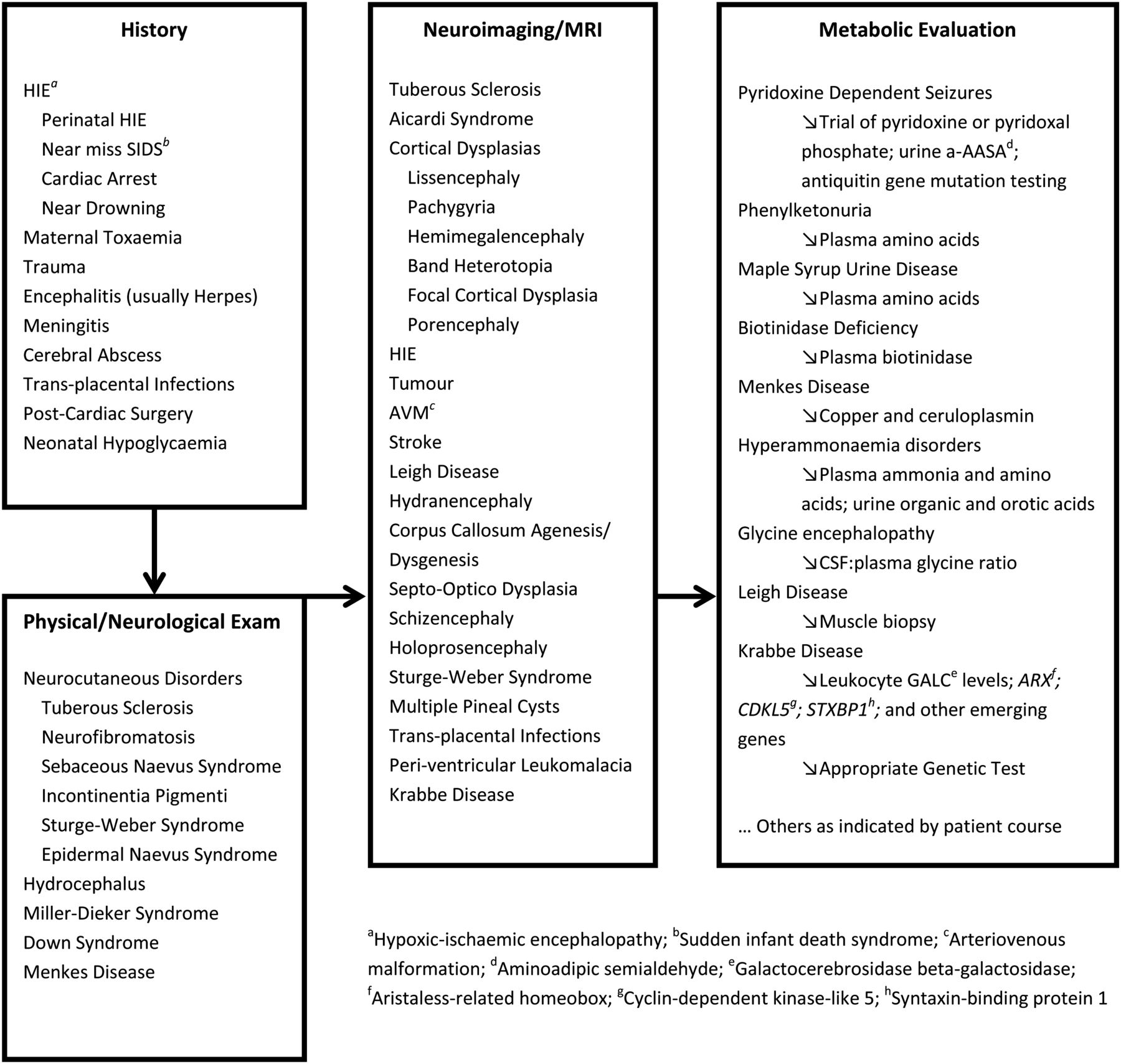{kind=link}
Aetiological clues and investigations
Seizures in infancy, at least after the neonatal period, have many potential causes and National Institute for Health and Clinical Excellence (NICE) guidance recommends early referral to an expert centre. There is no formulaic approach to investigating infantile epilepsy but there are some general principles. MRI is highly informative and is fundamental to the early assessment of aetiology. Subtler developmental abnormalities may be undetectable on early MRI scans because the process of brain myelination continues until about 30 months of age. Other clues include:
Symptoms and signs:
-
Severe startle reactions and hiccups (often elicited from the prenatal history as occurring in utero) are associated with glycine encephalopathy (non-ketotic hyperglycinaemia). This condition generally presents in the neonatal period with very severe seizures and a suppression-burst EEG (early myoclonic encephalopathy (see table 2)).
-
Hypopigmented skin lesions, for which a Woods’ lamp examination is required, are associated with tuberous sclerosis, which is strongly associated with focal seizures and with infantile spasms.
-
Sparse, steely or ‘kinky’ hair suggests Menkes disease, which is investigated by microscopic analysis of hair and for low levels of plasma copper and caeruloplasmin (although the blood tests are insensitive in early infancy).
-
Jitteriness and dystonic movements are often mistaken for epilepsy in infants presenting with glutaric aciduria type 1 (GA1). In GA1, there is usually macrocephaly and enlarged extra-axial cerebrospinal fluid (CSF) spaces, which can lead to a mistaken diagnosis of non-accidental head injury. In this and some other metabolic conditions, sodium valproate may worsen seizures by adverse effects on mitochondrial function, and treatment should include carnitine supplementation.18
-
Faltering growth and hypotonia suggest mitochondrial cytopathies such as Leigh syndrome.
Biochemistry:
-
Amongst the metabolic disorders that present with early myoclonic encephalopathy is molybdenum cofactor deficiency, which leads to functional deficit in the enzyme sulphite oxidase. Clues include low plasma uric acid and high urine xanthines and sulphites. Administration of cyclic pyranopterin monophosphate has recently been shown to be an effective treatment intervention, although it is not yet an approved treatment.19
-
Congenital microcephaly, seizures and developmental delay may be due to disorders of serine synthesis, generally detected on plasma amino acids and amenable to prenatal and postnatal supplementation with serine and glycine.
-
Biotinidase deficiency may mimic mitochondrial cytopathies, with developmental delay, episodes of encephalopathy and associated metabolic acidosis. It is usually associated with a severe rash, often with alopecia. Intractable epilepsy in infants is often treated with an empirical trial of biotin pending biotinidase assay results.
Electrophysiology:
-
In early infancy, EEGs with burst-suppression patterns are more likely to have structural brain lesions where seizures are tonic in semiology (Ohtahara syndrome) and more likely to have a metabolic cause where seizures are predominantly myoclonic (early myoclonic encephalopathy) (table 2).
Genetics:
-
Mutations in the STXBP1 (syntaxin-binding protein 1) gene affect neurotransmitter release and have been associated with Ohtahara syndrome and West syndrome.20
-
A history of seizures and learning disability affecting solely female family members suggests a mutation in the PCDH19 (protocadherin) gene, which has X-linked inheritance.21
-
Male infants with infantile spasms and/or myoclonic seizures may have mutations in the Aristaless-related homeobox gene. Other clinical features are variable and may include developmental delay, movement disorders, spasticity, abnormal genitalia and a spectrum of structural brain abnormalities that includes lissencephaly, midbrain malformations and agenesis of the corpus callosum.22
-
Female infants who have a Rett-like phenotype with early-onset seizures (the ‘Hanefeld variant’) and acquired microcephaly may have mutations in the CDKL5 (cyclin-dependent, kinase-like) gene.23 The clinical and EEG features can be distinctive.24 A similar phenotype is found with mutations in the FOXG1 (forkhead box G1) gene.25
-
Myoclonic encephalopathy may be associated with chromosomal disorders such as Angelman syndrome and chromosome 4p deletions.
-
Prolonged or hemi-convulsive seizures associated with fever should raise suspicions of abnormalities of neuronal sodium channel α-1 subunits, which are confirmed by mutation analysis of the SCN1A gene. This gene is associated with a range of conditions, including Dravet syndrome (table 2) and what was formerly considered to be ‘vaccine encephalopathy’.26
Metabolic conditions not to be missed
Pyridoxine and pyridoxal phosphate
Intractable seizures presenting in the neonatal period or early infancy are possibly due to rare but treatable metabolic diseases that should not be missed. These include pyridoxine-dependent epilepsy and pyridoxal phosphate-responsive epilepsy, which should be considered in any case of intractable epilepsy occurring under 2 years of age.27 Both conditions will respond to treatment with oral or nasogastric pyridoxal phosphate but this treatment is less well tolerated than oral pyridoxine—and neither is very well tolerated. The diagnosis should be considered as a differential when dealing with suspected hypoxic-ischaemic encephalopathy, which is relatively common, as the presentations can be similar.
-
Pyridoxine-dependent epilepsy is due to deficiency of α-AASA-dehydrogenase. Alpha-aminoadipic semialdehyde (α-AASA) is detected in blood and urine, with raised plasma pipecolic acid being a less sensitive marker. The biochemical abnormalities remain during treatment, so there is no reason for a trial of treatment to be delayed. Mutations of the antiquitin gene (ALDH7A1) confirm the genetic diagnosis. Seizures responsive to folinic acid are now considered to be the same condition.
-
Pyridoxal phosphate-responsive epilepsy is due to deficiency of the enzyme pyridox(am)ine phosphate oxidase and genetic confirmation is by testing of the PNPO gene. Plasma amino acid testing may show raised glycine and threonine, as might CSF. A more sensitive biochemical investigation is assay of CSF neurotransmitter metabolites, with low levels of pyridoxal-5′-phosphate levels, and possibly low levels of homovanillic acid and 5-hyroxyindole acetic acid.
Glucose transporter deficiency
Glucose transporter-1 deficiency has become increasingly recognised and is important to bear in mind as the associated seizures are generally intractable to conventional antiepileptic drug treatment but ketogenic diet is usually effective (though less effective if movement disorders are prominent). A combination of seizures, movement disorder and faltering head growth should trigger consideration of the diagnosis, but the epilepsy phenotype is being recognised as increasingly broad. In infants, features may also include apnoea and cyanosis. In older infants and children, the features may be much more subtle. For example, it has been described in patients diagnosed as having benign myoclonic epilepsy of infancy, myoclonic-atonic (‘myoclonic-astatic’) seizures or intractable absence-type seizures.28
Glucose transporter-1 deficiency is detected by measuring absolute CSF glucose levels and the ratio of CSF glucose to immediate pre-lumbar puncture venous glucose (ie, before the stress of lumbar puncture); and by mutations in the SCL2A1 gene. Neither of these diagnostic approaches is 100% sensitive, so case definition can be challenging.
Cerebral creatine deficiencies
It has recently been recognised that epilepsy in infancy or early childhood can be due to cerebral creatine deficiency.
-
Secondary to guanidinoacetate aminotransferase deficiency, this can be treated with creatine monohydrate and restriction of arginine. Other clinical features may include developmental delay, muscle hypotonia, extrapyramidal movements, abnormal eye movements and later, autistic and aggressive behaviour. Guanidinoacetate aminotransferase deficiency is detected by high levels of urine guanidinoacetate and absence of the normal creatine peak on magnetic resonance spectroscopy. An abnormal MRI signal in the globi pallidi is a less specific finding that has been reported in some cases.
-
Cerebral creatine deficiency secondary to mutations in the cerebral creatine transporter gene (SLC6A8) on the X-chromosome are not effectively treated with creatine supplementation. They seem to be associated with slightly later-onset epilepsy but the clinical phenotypes need fuller determination.
-
Deficiency of arginine:glycine amidinotransferase appears to be the least common form of cerebral creatine deficiency, with the main features being developmental delay and autism rather than epilepsy.
Delay, epilepsy and neonatal diabetes syndrome
The combination of developmental delay, epilepsy and neonatal diabetes is found with mutations in the potassium-ATP channel Kir6.2.29 This can present as infantile spasms. Neurological outcome can be improved by early treatment with oral sulphonylurea, whereas insulin treatment is ineffective and will not protect development.
Treatment of infantile epilepsy
Clearly, the first steps to effective treatment are correctly recognising epilepsy, seizure types and underlying causes. Treatment with antiepileptic drugs should not inhibit an appropriate search for treatable metabolic causes. Treatments are not discussed in detail here but a few learning points are noted.
Treatment guidelines
Guidelines on the treatment of epilepsy have been produced by the UK NICE and by the Scottish Intercollegiate Guidelines Network. The most recent NICE guidance30 (CG137) states that ‘the diagnosis and management of epilepsy within the first few years of life may be extremely challenging. For this reason, children with suspected epilepsy should be referred to tertiary services early, because of the profound developmental, behavioural and psychological effects that may be associated with continuing seizures’.
Sodium valproate
Sodium valproate is subject to national and cultural differences relating to perceived risks and use in infancy. It is a less commonly used first-line treatment in North America than in Europe and can be problematic in some situations. For example, in GA1 and in combination with the ketogenic diet, it can deplete carnitine. It is also problematic in mitochondrial disorders, and in particular, Alpers disease and mutations in the POLG1 (polymerase gamma) gene, where it can interfere with mitochondrial metabolism and precipitate liver failure.
Ketogenic diet
A ketogenic diet can be an effective intervention where antiepileptic drugs have been ineffective. The diet is modified in infants in order to minimise risks to growth. Its strongest indications are glucose transporter deficiency and pyruvate dehydrogenase (E1) deficiency, where the metabolic defects preclude brain utilisation of carbohydrates. However, it is contraindicated in pyruvate carboxylase deficiency, porphyria and defects of fatty acid oxidation.
Epilepsy surgery
Even in infancy, intractable epilepsy can be treated surgically. Indeed, early intervention is likely to reduce secondary epileptogenesis, and the maturing brain has a greater propensity for plasticity than the older brain, so there might be significant advantage associated with early referral and investigation.
Question 5: What is the prognosis?
Development and comorbidities
ILAE axis 5 comprises comorbidities, the associated cognitive, behavioural, emotional and developmental features that may be associated with the epilepsy. This corresponds with an important question that will be asked by parents and families of infants with epilepsy: ‘What is the prognosis for development?’ One of the great challenges facing the paediatrician is to reliably identify any underlying cause, which will strongly predict prognosis for the development of comorbidities, but also to appreciate where extensive metabolic and genetic investigation is unlikely to yield such an aetiological diagnosis given the current state of knowledge in this field.
When counselling families about prognosis for development, it is prudent to bear in mind case ascertainment bias. Early reports of cases with recently identified conditions tend to come from teaching centres, which generally have a more severe case mix. There can be period effects, with less severe cases being identified over time because investigations are performed more frequently and phenotypic spectra are recognised as being broader than previously imagined. Furthermore, in providing information about prognosis, it is also prudent to consider the effects over time of more effective treatment interventions, even if merely symptomatic or supportive treatments. For newer genetic conditions, information about longer-term prognosis is likely to be limited.
It is important to keep in mind the underlying motivation behind treating a child with epilepsy. It might not always be possible to stop seizures, in which case it is necessary to find balance between reasonable seizure control and unacceptable adverse effects associated with the treatments themselves.
Risks of sudden death?
One controversial issue is that of counselling risks of sudden unexpected death in epilepsy. Although nobody wishes to increase stress in an already stressful situation, NICE guidance encourages healthcare professionals to share and contextualise this information. The families of people who have suffered sudden death generally state that they would have preferred that information to have been offered before the event. Mortality from sudden unexpected death in epilepsy or other causes is higher in infants with epilepsy than in other children. For example, conditions such as Ohtahara syndrome, migrating epilepsy of infancy and Dravet syndrome have significant excess mortality in spite of appropriate treatments.
Sources of information and training
The websites of Epilepsy Action (http://www.epilepsy.org.uk/) and the Epilepsy Society (http://www.epilepsysociety.org.uk/) (formerly the National Society for Epilepsy) have very useful information for patients, families and professionals. Infantile seizures and epilepsies are covered during days 1 and 2 of the paediatric epilepsy training level 2 training course run by the British Paediatric Neurology Association.
Summary and key learning points
-
Epilepsies in infancy may be the manifestation of a genetic predisposition associated with a benign course and good prognosis for neurodevelopment. However, they may pose a challenging situation of ‘epileptic encephalopathy’, rare but potentially treatable metabolic conditions, or structural abnormalities with poor developmental outlook and intractable seizures. The 2001 ILAE 5-axis proposal provides a pragmatic framework for clinical diagnosis and management, although newer classification schemes are being developed. However, seizures in infancy are relatively rare and there are a wide range of underlying causes, some of which require specific treatments to avoid preventable neurodevelopmental damage. NICE guidance recommends early referral of cases to a tertiary centre.
Acknowledgments
ALL is treasurer of the British Paediatric Neurology Association, a not-for-profit organisation that runs the paediatric epilepsy training courses mentioned in this article.
References
Footnotes
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.