Article Text

Abstract
The expansion of the internet has resulted in widespread availability of medical information for both patients and physicians. People increasingly spend time on the internet searching for an explanation, diagnosis or treatment for their symptoms. Regarding rare diseases, the use of the internet may be an important tool in the diagnostic process. The authors present two cases in which concerned parents made a correct diagnosis of a lysosomal storage disorder in their child by searching the internet after a long doctor's delay. These cases illustrate the utility of publicly available internet search engines in diagnosing rare disorders and in addition illustrate the lengthy diagnostic odyssey which is common in these disorders.
Statistics from Altmetric.com
Introduction
The expansion of the internet has resulted in widespread public access to both scientific and non-scientific information. When confronted with a health complaint, people increasingly spend time on the internet searching for a diagnosis or treatment. Many physicians nowadays are confronted with patients or parents who present potential diagnoses and treatments for their complaints, which were retrieved from the internet. This behaviour undoubtedly influences patient/parent–physician relationship.1 2 The suggested diseases or remedies found on the internet may not be relevant to the patient's health issues, presenting rare diagnoses or treatment strategies that do not fall within the realm of conventional medicine for more common complaints and disorders. But sometimes these internet searches may reveal the correct diagnosis for rare diseases, which were not considered by the patients' physician. Here, we describe two cases where, after a long doctor's delay, concerned parents diagnosed a lysosomal storage disorder (LSD) in their child by searching the internet.
Case A
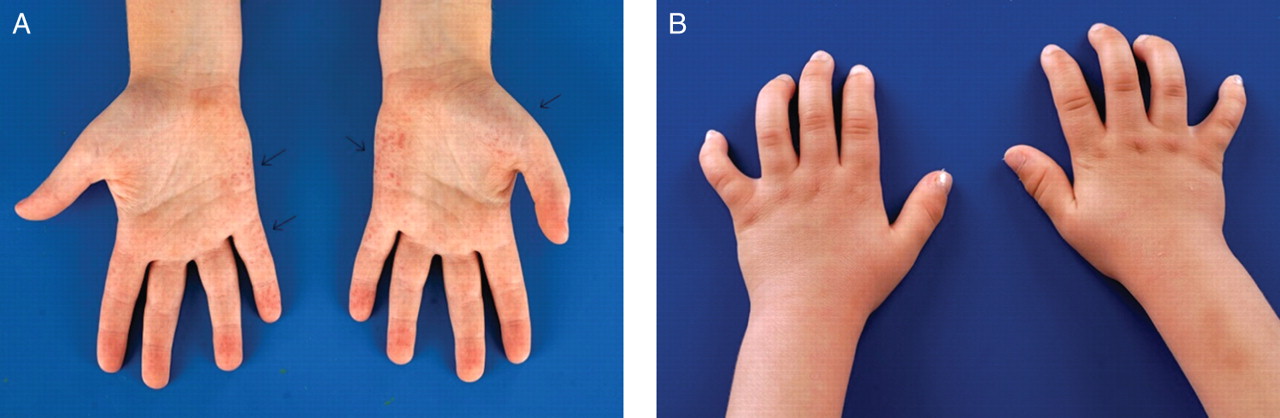
An 11-year-old boy was referred to our metabolic centre after his parents had suggested a diagnosis of Fabry disease to their paediatrician. From the age of 5 years he had suffered from severe episodic pains in the hands and feet, especially during exercise and fever. In addition, he had unexplained recurrent fever, joint aches and a skin rash on hands and feet. Three different paediatricians were consulted: a general paediatrician and two specialists in periodic fever syndromes and immunology, respectively. This led to the consideration of the following diagnoses: growing pains, juvenile idiopathic arthritis, hyper-IgD syndrome, tumour necrosis factor receptor-1-associated periodic syndrome and familial Mediterranean fever. Extensive laboratory tests were performed, but none of these diagnoses could be confirmed. Because symptoms persisted, the concerned parents initiated a search on internet, using ‘Google’ as search engine. They used the following search terms (in Dutch): unexplained recurrent fever, pain in feet and skin rash. Browsing the internet for several hours, they found a picture of a skin rash in a patient with Fabry disease. They immediately recognised the skin rash as similar to their child's, and after studying all available information, they concluded that their child might suffer from Fabry disease. On physical examination, angiokeratoma were present on the hands, feet, knees and hips (figure 1A). The consulted ophthalmologist found cornea verticillata. The diagnosis of the LSD Fabry disease was confirmed by enzyme analysis. There were no signs of renal and cardiac involvement, as often seen at this age. He was started on enzyme replacement therapy (ERT) within the scope of an early intervention trial (FIELD study, NCT00701415) .
{kind=link}
(A) Patient A: angiokeratoma on the palmar side of the hands. Right hand ulnar side and left hand ulnar side and thenar. (B) Bowed fingers on both hands on patient B.
Fabry disease is an X linked LSD caused by a deficiency of the enzyme α-galactosidase A (αGAL-A). The main substrate of αGAL-A, globotriaosylceramide (Gb-3), accumulates in endothelial cells and other cell types. In childhood, Fabry disease is characterised by severe episodic pains in the hands and feet (acroparaesthesia), anhydrosis and angiokeratoma. In adult life, disease progression causes renal, cardiac and neurological morbidity, and a reduced life-span. Since 2001, therapy has become available as ERT. Treatment with ERT reduces tissue Gb-3 and may stabilise renal disease and other disease features.3
Case B
A boy, aged 16 months, was referred to a metabolic centre, after his mother suspected that he might have mucopolysaccharidosis type I (MPS I). Born prematurely at 33 weeks (1790 g) from non-consanguineous parents, the boy needed continuous pulmonary airway pressure during his first 3 days of life. He failed his neonatal hearing tests. In the following 3 months, he was admitted several times because of excessive crying.
At the age of 3 months, he was seen because of macrocephaly. A mild hydrocephalus without signs of high pressure was diagnosed on MRI. A wait-and-see policy was adopted. At the age of 5 months, a one-sided inguinal hernia was corrected.
The child was referred to a clinical geneticist at the age of 7 months, primarily for investigation of myotonic dystrophy type 1, an autosomal dominant disorder that had been diagnosed in his father. Apart from the possibility of myotonic dystrophy, the geneticist was also asked to consider other diagnoses, in view of his symptoms. No alternative diagnosis was suggested by the clinical geneticist.
The child had recurrent upper airway infections during his first year of life and was noted to snore significantly during sleep. At the age 10 months, an adenectomy was performed, grommets were placed, and he received hearing aids. His parents noticed kyphosis, changing facial features, bowed fingers (figure 1B), an umbilical hernia and slowing of psychomotor development. This latter observation resulted in cognitive testing, which revealed an IQ of 72 (−1 to −2 SD). Numerous visits to the paediatrician did not lead to a diagnosis for the plethora of signs and symptoms. The mother, who was anxious to know what was wrong with her child, repeatedly searched the internet for a diagnosis. One day, she entered ‘bowed fingers’ (in Dutch) into the search engine ‘Google,’ and the first hit was a Belgium website on lysosomal storage disorders. Studying this site, which showed pictures of children with MPS I, the mother immediately recognised the specific facial features of her child and thought that MPS I, Hurler phenotype, could be a likely diagnosis. Confronted with this information, the paediatrician requested appropriate enzymatic studies, confirming the diagnosis. The patient received successful haematopoietic stem cell transplantation (HSCT) at the age of 19 months, after a treatment period with ERT.
MPS I is an autosomal recessive LSD caused by a deficiency of the enzyme α-L-iduronidase. Accumulation of glycosaminoglycans leads to cellular and organ dysfunction. Three phenotypes are recognised: the severe Hurler, the intermediate Hurler–Scheie and the attenuated Scheie syndrome. Patients with the Hurler syndrome have marked cognitive delay, coarse facial features, corneal clouding, hearing imparement, hepatosplenomegaly, umbilical and inguinal hernias, and orthopaedic cardiac and respiratory problems in early childhood. Without treatment, life expectancy is limited. Patients with the Scheie syndrome have normal intelligence and survive into adulthood, although they may experience significant morbidity such as restricted joint mobility, carpal tunnel syndrome, skeletal dysplasia, cardiac involvement and pulmonary dysfunction. HSCT may preserve mental function if performed at an early age, generally before the age of 2 years. In addition, ERT has become available since 2003 for the treatment of the non-neurological complications in MPS I.4
Conclusion
These cases illustrate the utility of publicly available internet search engines in diagnosing rare disorders, such as LSDs. They also demonstrate the lengthy diagnostic odyssey that often occurs in these disorders. A prolonged diagnostic odyssey is more devastating if an early diagnosis is of vital importance for the efficacy of treatment. This is indeed the case for both presented disorders. ERT in patients with Fabry disease is less efficacious when started when the disease already progressed.5 6 In patients with the severe Hurler phenotype of MPS I, HSCT is preferably done as early as possible, before significant involvement of the central nervous system (CNS).7 Early HSCT improves the chances both for engraftment8 and for prevention of CNS complications. Given the rare nature of these disorders, many paediatricians will never see a patient with these diseases and will not recognise it, and subsequently a diagnosis is unlikely to be made. Faced with an undiagnosed disorder in their child, many parents will start roaming the internet. The presented cases show that this may result in a final diagnosis. It has been previously reported that the search engine ‘Google’ can be a useful diagnostic tool for both physicians9 10 and non-physicians,11 12 especially in difficult diagnostic cases. Tang and Ng selected 26 complex cases presented in the New England Journal of Medicine: in 58% of the cases, ‘Google’ revealed the correct diagnosis.10 Siempos et al followed the same strategy in non-physicians, which resulted in a correct diagnosis in 22.1% of the cases.12 The difference in success rate could be because non-physicians may find it more difficult to interpret quality and reliability of information on the internet.2 The search terms used by physicians and non-physicians might also explain these differences. The parents of the presented cases did not have a medical background and were of average education. Apart from the recognition of symptoms leading to diagnosis of a disorder, as illustrated here and elsewhere,10,–,12 frequent searches on the internet may also lead to anxiety and medical overconsumption, also known as cyberchondria.2 13 14
Nowadays, with easy internet access, physicians should acknowledge the transformation of patients from passive recipients to active consumers of health and should be open to discuss the value of the information offered by the patients. Finding the right balance between patient-suggested ‘internet-made-diagnosis’ and medical overconsumption can be challenging and is a relatively new dilemma in medicine. Finally, the presented cases demonstrate that physicians, when confronted with unrecognised combinations of signs and symptoms, should use internet searches themselves as part of their diagnostic strategy to prevent unacceptable diagnostic delay.
References
Footnotes
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Patient consent Obtained from the parents.